




















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Interstitielle Lungenerkrankungen
Früh erkennen, früh therapieren
18. April 2025
Interstitielle Lungenerkrankungen (ILD) sind eher selten, in ihrer Gesamtheit betreffen die fibrosierenden Formen bei einer Prävalenz von ca. 100/100.000 ca. eine von 1.000 Personen. Die gesamte Gruppe der interstitiellen Lungenerkrankungen umfasst über 100 verschiedene Diagnosen, wobei die mit Abstand häufigsten folgende sind:
- Idiopathische Lungenfibrose. Betrifft meist ältere Menschen männlichen Geschlechts mit Raucher- bzw. Schadstoffexpositionsanamnese. Klassisch zeigt sich ein Usual-interstitial-Pneumonia-(UIP-)Muster in der CT (Abb.) und in der Histologie. Fast immer kommt es zu einem fortschreitend vernarbenden Verlauf mit ungünstiger Prognose, das mediane Überleben liegt bei weniger als 5 Jahren.
- ILD im Rahmen von Autoimmunerkrankungen. Am häufigsten bei systemischer Sklerose, rheumatoider Arthritis und Myositiden, verbreiteter bei Frauen und radiologisch meist mit nichtspezifischer interstitieller Pneumonie (NSIP) im CT.
- Exogen allergische Alveolitis (EAA). Entsteht durch repetitive allergische Typ-III/IV-Reaktion auf inhalierte organische Antigene (z. B. Vogelantigene, Schimmelpilzstäube). Die akute Form ist durch Fieberschübe und Atemnot, die chronische Form durch Ausbildung einer Fibrose charakterisiert. Prognose bei Antigen-Identifikation und Elimination gut, bei fehlendem Nachweis und fortschreitender Fibrose schlecht.
- Sarkoidose. Charakterisiert durch das Auftreten von epitheloidzelligen Riesenzellgranulomen in den mediastinalen/hilären Lymphknoten, der Lunge und auch – je nach Befallsmuster seltener – in anderen Organen. Bessere Prognose als die meisten anderen ILD, wobei auch bei der Sarkoidose ca. 10 % der Fälle einen ungünstigen, progredient fibrosierenden Verlauf nehmen.
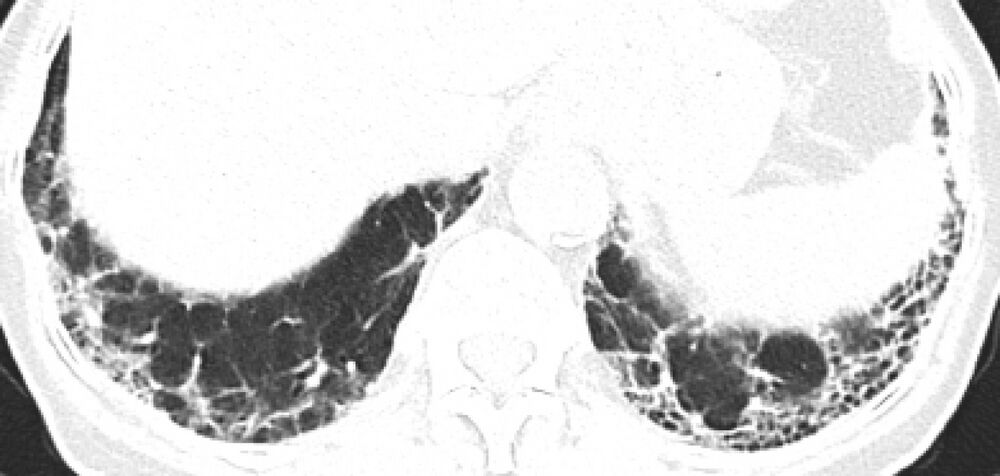
Abb.: Computertomografiebild einer Patientin mit klassischem UIP-Muster. Man sieht deutlich (linksbetont) die Honigwabenzysten, daneben auch retikuläre Veränderungen.
Diagnose von ILD
Da ILD selten sind und die Präsentationssymptome meist uncharakteristisch bleiben, besteht oft eine lange Verzögerung der Diagnosestellung, was die Prognose verschlechtert. Ein wichtiger und einfacher Teil der Untersuchung ist die Auskultation: Bei einem Großteil der ILD-Patient:innen mit fibrosierenden Erkrankungen zeigt sich das charakteristische spätinspiratorische Knisterrasseln (Sklerosiphonie).
Bei Verdacht sollte jedenfalls eine CT des Thorax erfolgen, die dann meist entscheidende Hinweise gibt. Als Faustregel gilt, dass das UIP-Muster mit Traktionsbronchiektasen und Honeycombing, egal bei welcher ILD-Entität es auftritt, immer eine schlechte Prognose hat. Bei allen ILD, insbesondere aber wenn ein NSIP-Muster vorliegt, sollte immer nach einer zugrundeliegenden Autoimmunerkrankung gesucht werden. Dies muss einerseits klinisch erfolgen, andererseits sollte bei allen ILD-Patient:innen bei der Erstabklärung ein autoimmunologisches Basisscreening mit ANA, ANCA, Rheumafaktor, CCP und Myositisantikörpern stattfinden. Eine Lungenbiopsie wird heutzutage nur noch selten per videoassistierter Thorakoskopie von Chirurg:innen, sondern viel öfter über eine transbronchiale periphere Kryobiopsie von Pneumolog:innen durchgeführt, was als weniger invasiv bei dennoch guter diagnostischer Ausbeute gilt.
Therapie von ILD
In der Pathogenese der ILD gibt es ein Kontinuum von fortschreitender irreversibler Vernarbung einerseits und aktiver Entzündungsaktivität andererseits. Es gibt Extremfälle wie die idiopathische Lungenfibrose als klassische fortschreitendfibrotische Erkrankung oder die Sarkoidose und die akute EAA, die stark entzündlich auftreten und in frühen Stadien unter antiinflammatorischer Therapie oft vollständig reversibel sind.
Zur Therapie inflammatorischer ILD gibt es zahllose immunsuppressive/immunmodulierende Substanzen, wobei neben systemischen Steroiden (Dauertherapie vermeiden!) heutzutage vor allem Mycophenolat-Mofetil, Methotrexat und Rituximab eingesetzt werden. Antifibrotische Therapien gibt es erst seit ca. 10 Jahren in Form der Medikamente Pirfenidon und Nintedanib, die über verschiedene Wege die Fibroblasten und somit die fortschreitende Vernarbung hemmen. Wichtig ist, dass diese Medikamente bei fortschreitenden ILD möglichst früh zum Einsatz kommen sollen, um einen weiteren Lungenfunktionsverlust zu verhindern und noch möglichst lange eine gute Lebensqualität zu ermöglichen. Eine Heilung von fibrosierenden ILD ist heute weiterhin nicht möglich, auch eine Verbesserung gelingt medikamentös vielfach nicht – das Ziel ist weiterhin die Stabilisierung. Patient:innen unter 70 Jahren und ohne schwere Komorbiditäten sollten auch bezüglich einer Lungentransplantation evaluiert werden, die Erstvorstellung an einem Transplantationszentrum sollte nicht zu spät erfolgen.
Ausblick
Aktuell ist das Hauptziel die frühe Detektion und Therapie von ILD, um die Lebensqualität bei diesen meist ungünstig verlaufenden Krankheiten möglichst lange zu erhalten. Die absehbare Zukunft wird neue Medikamente bringen, insbesondere die Substanz Nerandomilast, für die bereits positive Studienergebnisse vorliegen und die sowohl an entzündlichen als auch an vernarbenden Mechanismen angreift.
Praxismemo
- Die Symptomatik ist oft unspezifisch: Belastungsdyspnoe und Husten können auf eine ILD hinweisen.
- ILD bei Autoimmunerkrankungen werden mit immunsuppressiven bzw. immunmodulierenden Substanzen behandelt.
- Bei fibrosierenden ILD hemmen Pirfenidon und Nintedanib die fortschreitende Vernarbung der Lunge.

Autor:
Priv.-Doz. Dr. David Lang, PhD
Priv.-Doz. Dr. David Lang, PhD
Universitätsklinik für Innere Medizin 4 – Pneumologie, Kepler Universitätsklinikum, Linz

Ursprünglich erschienen:
AEK 08|2025
AEK 08|2025
AWARENESS FÜR ILD
Wichtig ist, das Bewusstsein von Ärzt:innen zu schärfen, dass Belastungsatemnot, Husten und eventuell ein „nicht ganz sauberes“ Thoraxröntgen insbesondere bei Menschen mit Risikofaktoren wie Nikotinabusus, Autoimmunerkrankungen oder speziellen inhalativen Expositionen schon den Anfang einer ILD darstellen können.
INTERDISZIPLINÄRE ABKLÄRUNG
Alle Befunde sollten interdisziplinär von einem ILD-Board besprochen werden, bei dem im besten Fall Pneumologie, Rheumatologie, Radiologie und Pathologie gemeinsam Diagnosen stellen und Therapieentscheidungen treffen. Patient:innen mit hochgradigem Verdacht auf ILD sollten daher in ILD-Zentren mit ILD-Board abgeklärt werden.
INFLAMMATION UND FIBROSE
Insbesondere bei schon länger andauerndem Krankheitsverlauf liegen häufig sowohl inflammatorische als auch fibrosierende Anteile nebeneinander vor. Verschiedene Biomarker aus Klinik, Labor, Histologie und Radiologie können helfen, die vordergründige Komponente zu identifizieren und zu therapieren.
TIPPS FÜR DIE PRAXIS
- Bei unklarem Husten und/oder Belastungsdyspnoe an ILD denken! Im Verdachtsfall auskultieren und zu Thorax-CT zuweisen.
- Diagnose und Therapieeinleitung sollten durch Spezialambulanzen nach Besprechung im multidisziplinären ILD-Board erfolgen.
- Auch geringe interstitielle Auffälligkeiten in der CT können relevant werden. Zuweisung an eine Spezialambulanz erwägen!
- Risikofaktoren für einen rasch fortschreitenden fibrosierenden Verlauf: UIP-Muster, Raucheranamnese, erhöhte Blutmonozyten, hohe entzündliche Aktivität.
- Bei Fortschreiten einer Fibrose radiologisch, funktionell oder klinisch: antifibrotische Medikamente Pirfenidon (IPF) oder Nintedanib (alle fortschreitend fibrosierenden ILD) erwägen!
WAS PATIENT:INNEN WISSEN WOLLEN
Was kann ich zusätzlich tun, um meine Lungenfibrose günstig zu beeinflussen?
Absolute Nikotinkarenz, körperliche Aktivität – am besten in Form einer Lungenrehabilitation –, ausgewogene proteinreiche Ernährung, idealerweise nach diätologischer Evaluation zur Verhinderung einer pulmonalen Kachexie.
Ich habe rheumatoide Arthritis mit Lungenfibrose. Warum wird meine Lunge schlechter, obwohl es mir unter Therapie gut geht?
Leider gibt es immer wieder Fälle, bei denen Lungen- und Gelenkmanifestationen nicht Hand in Hand gehen. Insbesondere eine bereits fortgeschrittene Lungenfibrose kann „von sich aus“ immer schlechter werden. Trotzdem sollte die Autoimmunerkrankung gut eingestellt sein, da eine hohe Rheumaaktivität meist auch schlecht für die Lunge ist.
Literatur beim Verfasser
Bildnachweis
Vorschaubild: © Iona – stock.adobe.com