





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
ÖGN 2012: Spezifische Diäten als Therapie neurologischer Erkrankungen
27. April 2012
Morbus Refsum
Es handelt sich um eine extrem seltene Lipidstoffwechselerkrankung (Phytansäurespeicherkrankheit), die autosomal rezessiv vererbt wird. 1946 erfolgte die Erstbeschreibung durch Sigvald Refsum als „Heredopathia atactica polyneuritiformis“ bei einer norwegischen Familie mit der klinischen Trias von Retinitis pigmentosa, Polyneuropathie und Ataxie. 1963 gelang der biochemische Nachweis der Phytansäurespeicherung in Gewebe und Blut. Für diesen „inborn error of metabolism“ sind mehrere Gendefekte bekannt, seit 1997 ist für einzelne davon ein molekulargenetischer Nachweis in spezialisierten Labors möglich. In den Zellorganellen namens Peroxisomen fehlt ein spezifisches Enzym, das die Phytansäure oxidiert.
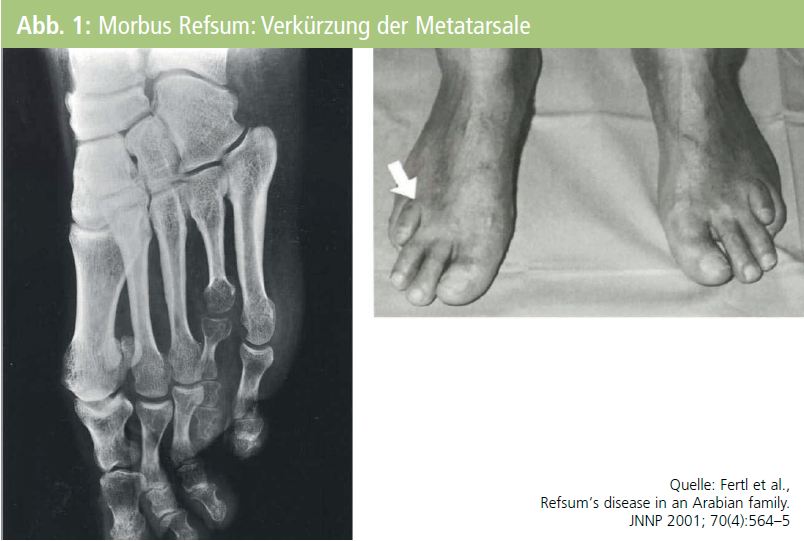
Der angeborene Mangel oder Dysfunktion des Enzyms Phytanoyl-Coenzym-A-Hydroxylase (PAHX) führt zu einem gestörten Abbau der rein exogen zugeführten Phytansäure, selbst wiederum ein Abbauprodukt des Pflanzenfarbstoffs Chlorophyll. Die mit der Nahrung aufgenommene Phytansäure lagert sich dann in der Retina, im peripheren Nervensystem, im Zentralnervensystem und im Herzmuskel an und führt dort zu progredienten Funktionsstörungen. Neben oft angeborenen typischen Skelettdeformitäten1 (Abb. 1) zeigen sich dann erworbene Störungen: progrediente Nachtblindheit und Tunnelblick durch Retinitis pigmentosa sowie in der 2. bis 3. Dekade eine progrediente distal betonte, zuerst rein sensible Polyneuropathie mit strumpfförmiger Hypästhesie.
In der Klassifikation der hereditären sensomotorischen Neuropathien wurde deshalb der Morbus Refsum als Typ IV klassifiziert. Die Ataxie entsteht wohl eher durch die gestörte Propriozeption als durch zerebelläre Veränderungen. Kernspintomographisch lässt sich initial im ZNS keine Veränderung nachweisen. Uns gelang etwa der erstmalige Nachweis dieser Erkrankung in einer Familie arabischer Herkunft im Jahr 2001. Eine seltene Variante ist der infantile Morbus Refsum, bei dem schon der Säugling Gedeihstörungen, sensoneurale Hörminderung und Hepatomegalie zeigt. Diese Kinder haben eine hereditär bedingte Störung der Biogenese der Peroxisomen, analog zu den Lipidspeicherkrankheiten der Adrenoleukodystrophie, Zellweger-Syndrom und der rhizomelischen Chondrodysplasie.
Westminster-Diät: Als Abbauprodukt von Chlorophyll ist die Phytansäure, eine langkettige, gesättigte und verzweigte C20-Fettsäure, in allen Nahrungsmitteln, die von wiederkäuenden Tieren oder Fisch stammen, enthalten. Fische und Wale fressen Algen und Krill, der besonders chlorophyllhaltig ist. Verminderte alimentäre Zufuhr von phytansäurehaltigen Produkten führt nachweislich zu einem geringeren Phytansäurespiegel im Serum und konsekutiv auch im Gewebe. Es gibt eine spezifische Diät (Tab. 1), die im Wesentlichen auf Fleisch von Wiederkäuern, Milchprodukte und tierische Öle verzichtet. Definitiv verboten sind Fleischprodukte von Rind, Schaf und Wild sowie Fisch und sämtliche Milchprodukte, Milch und tierische Fette. Grünes Gemüse und Obst sind unbedenklich, weil das dort enthaltene Chlorophyll im menschlichen Organismus nicht zu Phytansäure abgebaut wird.
PatientInnen mit adultem Morbus Refsum, die diese Diät langfristig einhalten, können eine Progression von Polyneuropathie und Retinopathie vermeiden. Langzeitdaten aus Großbritannien zeigen, dass die spezifische Diät als einzige Therapie des Morbus Refsum stark wirksam ist2. Wenn ein/eine PatientIn mit Morbus Refsum allerdings rasch Gewicht verliert oder im Zuge einer schweren Allgemeinerkrankung mit katabol bedingtem Fettgewebsabbau Phytansäure mobilisiert, kann eine krisenhafte Verschlechterung eintreten, die eine Akuttherapie mit Plasmapherese notwendig macht.
Pseudotumor cerebri – idiopathische intrakranielle Hypertension (IIH)
Der Pseudotumor cerebri ist eine relativ häufige Erkrankung vorwiegend von jungen, adipösen Frauen, bei der aus unbekannter Ursache eine Erhöhung des Druckes im intrathekalen Kompartiment besteht. Die Prävalenz beträgt 1–3 Personen/100.000/Jahr in der Durchschnittsbevölkerung und steigt auf 19/100.000 bei Übergewichtigen. Durch die weltweit zunehmende Adipositasepidemie wird in Zukunft weiter mit einer Prävalenzsteigerung der Erkrankung zu rechnen sein. Hauptsymptome sind holokraniale Cephalea und Visusstörungen durch ein Ödem der Sehnervenpapille. Zerebrale Bildgebung und Neurostatus sind üblicherweise unauffällig, Funduskopie oder optische Kohärenztomographie zeigen eine bilaterale Papillenschwellung. Die Diagnose wird durch Lumbalpunktion mit erhöhtem intrathekalem Druck bei sonst unauffälliger Zusammensetzung des Liquor cerebrospinalis gestellt. Unerkannt kann die Erkrankung letztlich zur Erblindung führen. Es gibt keine Therapieleitlinien, 2005 konnte ein Cochrane-Review etwa keine einzige randomisierte Interventionsstudie bei Pseudotumor cerebri finden. Mittlerweile gibt es zu einzelnen Pharmaka (Azetazolamid, Topiramat3) erste Evidenz. Neben medikamentösen und operativen Therapieverfahren, die aber erst als Ultima Ratio eingesetzt werden, gibt es nun erstmals Erfahrungen mit einer diätetischen Behandlung.
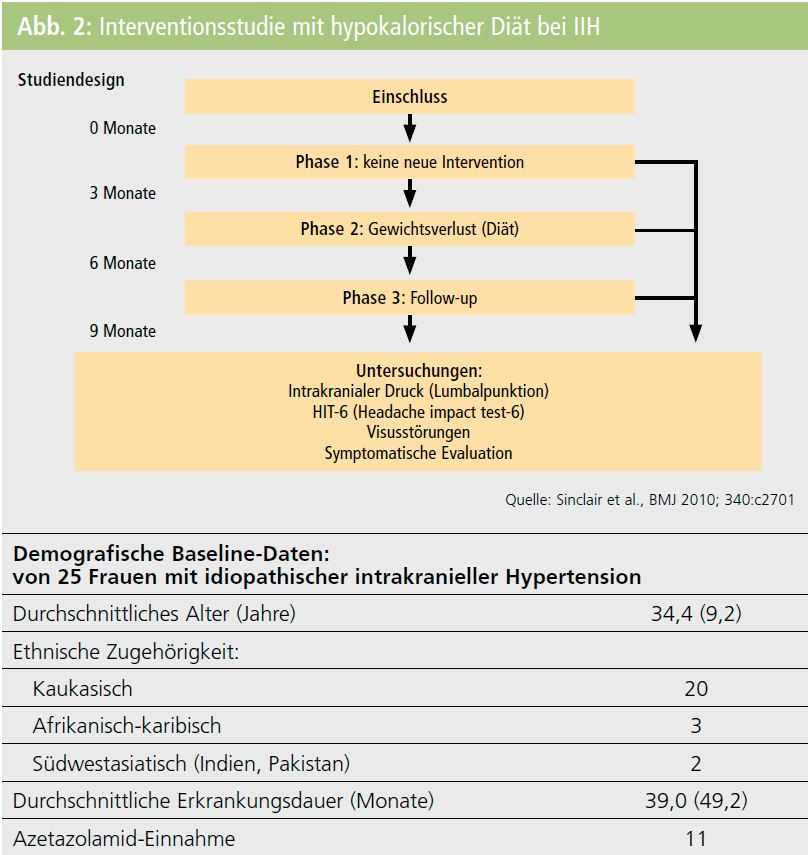
Diätetische Behandlung: 2010 wurde in Birmingham, Großbritannien, eine prospektive Kohortenstudie im A-B-A-Design an 25 jungen Frauen mit IIH und einem Durchschnittsgewicht von 101 kg durchgeführt, wobei die Patientinnen insgesamt 9 Monate engmaschig überwacht wurden und drei Mal eine Liquorpunktion durchgeführt wurde. Die ersten 3 Monate waren eine Baseline-Phase ohne Intervention, die zweite 3-Monats-Periode bestand in diätetischer Behandlung mit einer hypokalorischen Trinknahrung (425 kcal pro Tag) und wöchentlichem ärztlichem Monitoring, gefolgt von einer 3-monatigen Nachsorgephase. Während der diätetischen Interventionsphase wurden die Patientinnen wöchentlich gesehen und die Compliance labormäßig überprüft. In der Interventionsphase verloren die Patientinnen durchschnittlich 15,6 kg und der Liquordruck wurde erheblich reduziert4 (Abb. 2). Weiters führte das strikte Einhalten der hypokalorischen reinen Trinknahrung zu einer deutlichen Reduktion von Kopfschmerzhäufigkeit und -intensität und des Analgetikakonsums. Ebenso konnte die Abnahme der Schwellung an den Sehnervenpapillen eindeutig dokumentiert werden. Die Behandlungseffekte blieben im Nachsorgezeitraum stabil. Diese Studie zeigt erstmals einen gangbaren diätetischen Weg für dieses sekundäre Kopfschmerzleiden auf, weitere Erfahrungen zu dieser Behandlungsmethode werden mit Spannung erwartet.
Zöliakie
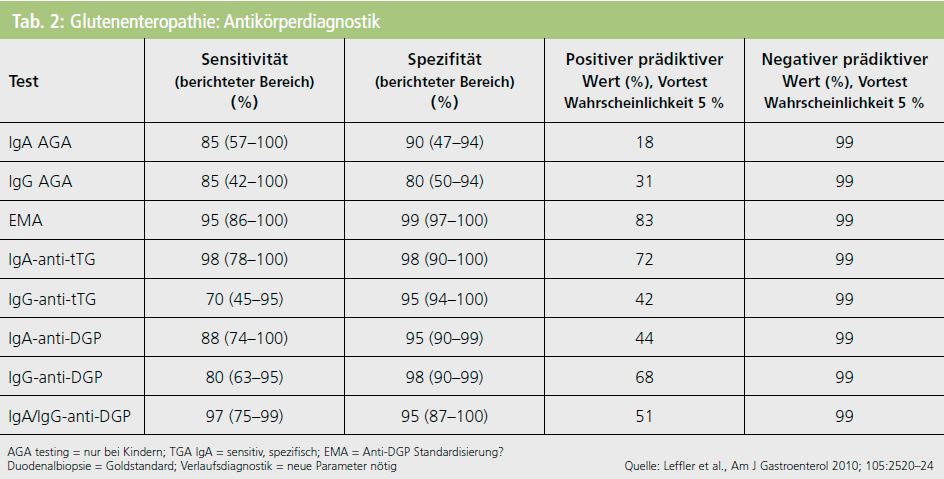
Die Zöliakie (Glutenenteropathie) ist in unseren Breiten mit 1 % Prävalenz eine häufige Erkrankung. Es handelt sich um eine Autoimmunerkrankung mit einer hohen genetischen Prädisposition (in 80 % HLA-Typ DQ2 oder DQ8), bei der eine Unverträglichkeit von Klebereiweiß (Gluten) besteht. Glutenproteine finden sich in verschiedensten Getreidesorten und deren Verarbeitungsprodukten (Mehl, Gebäck, Teigwaren, Saucen). Die enterale Aufnahme von Gluten führt zu einer T-Zell-vermittelten Entzündungsreaktion im Dünndarm mit Zottenatrophie und Malabsorption. In die Zirkulation gelangen mehrere gastrointestinale und auch antineuronale Autoantikörper. Serologisch können spezifische Autoantikörper gegen das gewebegebundene Enzym Transglutaminase (TGA) nachgewiesen werden. Typischerweise wird im Darm das Isoenzym TG2 exprimiert, während im Nervensystem vor Kurzem die Isoform TG6 gefunden wurde. Die früher häufig zum Screening eingesetzten Anti- Gliadin-Antikörper der Klasse IgG oder IgA (AGA) werden wegen geringer Sensitivität und Spezifität heute in der Diagnostik nicht mehr eingesetzt.
Symptomatik: Die Symptome der Glutenunverträglichkeit sind jedoch äußerst variabel in der klinischen Ausprägung und im Manifestationsalter: Es gibt gastrointestinale, dermatologische und neurologische Symptome, aber auch völlig asymptomatische Betroffene. In der Gastroenterologie kennt man Gedeihstörungen beim Säugling nach dem Abstillen, fluktuierende Durchfälle und Bauchschmerzen im Kindes- und Jugendalter und schwere Malabsorptionssyndrome mit Haarausfall, Osteoporose und Marasmus.
Neurologische Symptome treten meist vor gastrointestinalen Beschwerden oder völlig ohne solche beim Erwachsenen auf und sind nur im ersten Jahr nach Manifestation unter spezifischer Diät reversibel5. Dies stellt NeurologInnen vor eine besondere diagnostische Herausforderung, weil im Einzelfall bei zöliakietypischen aber ätiologisch ungeklärten Symptomen/Syndromen nach einer Glutenunverträglichkeit gezielt gesucht werden muss (Tab. 2).
Auch gibt es eine dermatologische Manifestation der Glutenunverträglichkeit, die Dermatitis herpetiformis mit Bläschen bildenden Exanthemen an Knie und Ellbogen mit Juckreiz. Diese PatientInnen exprimieren Autoantikörper gegen die Isoform TG3 im Serum.
In der Gastroenterologie erfolgt die Diagnosestellung über ein serologisches Screening (Bestimmung von gesamt IgA und Anti-TGAIgA; Tab. 2) und als Bestätigungstest eine Dünndarmbiopsie mit typischem Befund. Die Dünndarmhistologie wird nach MARSH in drei Schweregrade klassifiziert und zeigt eine Zunahme intraepithelialer Lymphozyten, Kryptenhyperplasie und letztlich Zottenatrophie.
Nach etablierter Diagnose erfolgt die Behandlung mit einer lebenslangen spezifischen glutenfreien Diät6. Vermieden werden müssen sämtliche Getreideprodukte (Roggen, Weizen, Gerste, Hafer, Dinkel, Hirse), aber auch Bier, das ebenso glutenhaltig ist. Bei guter Adhärenz zur glutenfreien Diät remittieren die gastrointestinalen Symptome, die Darmschleimhaut erholt sich und auch der Titer der Autoantikörper fällt unter die Nachweisgrenze. Einschulung in die glutenfreie Diät und Verstärkung der Adhärenz sind Domänen von Diät- und ernährungsmedizinischen BeraterInnen. Zur Unterstützung bei der Diättherapie wurden in Deutschland und Österreich schon vor Jahrzehnten große Selbsthilfegruppen gegründet, die mit Fachwissen, Diätberatung und jährlichen Fachtagungen die Betroffenen unterstützen.
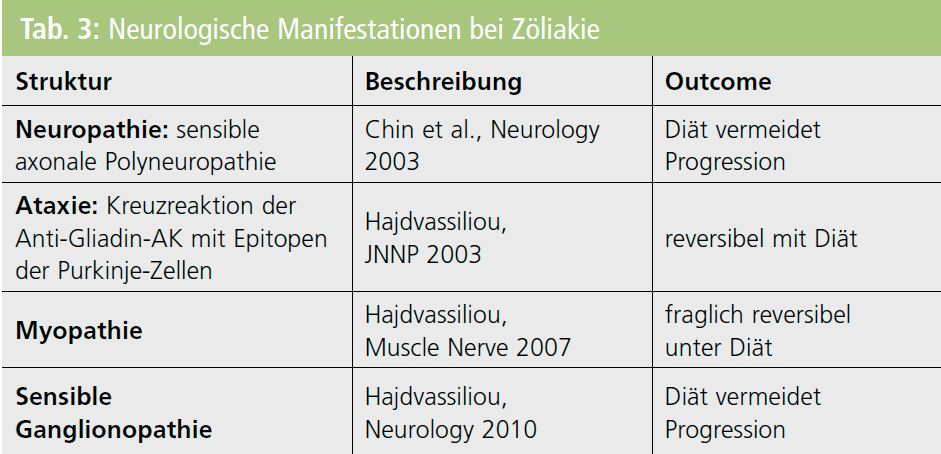
Neurologische Beeinträchtigungen: Zusammenhänge zwischen Zöliakie und neurologischen Beeinträchtigungen wurden bereits in den 1960er-Jahren beschrieben, sind aber teilweise noch immer unklar bzw. obskur (Tab. 3). Unbestritten sind die Assoziationen zwischen Zöliakie und zerebellärer Ataxie (Glutenataxie) sowie sensibler symmetrischer Polyneuropathie (Glutenneuropathie). Neuere Evidenz berichtet von Assoziation mit sensibler Ganglionopathie, Myopathie, Epilepsie und kognitiven Beeinträchtigungen. Eine besondere Bedeutung bekommt diese Frühdiagnostik, weil Ataxie und sensible Polyneuropathie durch strenge diätetische Behandlung nur in der Frühphase reversibel sind.
Neuropathologisch gibt es wenig Evidenz für die Pathophysiologie der Zöliakieveränderungen am zentralen und peripheren Nervensystem. Neben Vitaminmangelerscheinungen sind sicherlich auch autoimmun bedingte Entzündungsmechanismen beteiligt. Am Kleinhirn kommt es zum Verlust der Purkinjezellen mit zerebellärer Atrophie, an den Hintersträngen zur Demyelinisierung und vereinzelt im Zentralnervensystem auch zur Lyawomit rein extraintestinale Symptome bei Glutenintoleranz mit normalem bioptischem Dünndarmbefund gemeint sind. Diese PatientInnen zeigen zwar typische zirkulierende Antikörper, aber keine intestinale Pathologie. Wenn sich dann typische neurologische Symptome unter glutenfreier Diät bessern, so ist eine Glutensensitivität gegeben.
Wegen der Häufigkeit unspezifischer, aber ätiologisch ungeklärter neurologischer Symptome und der gleichzeitig hohen Prävalenz der Zöliakie besteht hier gelegentlich ein diagnostisches Dilemma. Für die neurologische Diagnostik von Glutensensitivität gibt es einen Vorschlag einer britischen Arbeitsgruppe7. Eine zentrale Rolle spielt der Nachweis von Anti-TG2-IgA-Antikörpern, die eine hohe Sensitivität und Spezifität für neurologische Komplikationen bei Zöliakie haben. Diese spezifischen Antikörper gegen TG2 – das Autoantigen bei Zöliakie – finden sich im Serum, im gastrointestinalen Stroma und auch rund um zerebrale Arteriolen. Der anti-TG2-IgA-Nachweis im Serum steht aber keineswegs überall in der entsprechenden Qualität zur Verfügung. Möglicherweise kann auch bald ein zuverlässiger Essay für Antikörper gegen TG6, das exklusiv im Gehirn exprimiert wird, entwickelt werden. Eine umfassendere wissenschaftliche Vertiefung in die Zusammenhänge zwischen der hereditär bedingten Autoimmunerkrankung Zöliakie und Manifestationen am peripheren und zentralen Nervensystem wäre äußerst wünschenswert.
1 Fertl E, Földy D, Vass K et al., Refsum´s disease in an Arabian Family. JNNP 2001; 70(4):564–5.
2 Baldwin EJ, Gibberd FB, Harley C et al., The effectiveness of long-term dietary therapy in the treatment of adult Refsum disease. JNNP 2010; 81:954–57.
3 Finsterer J, Földy D, Fertl E, Topiramate resolves headache from pseudotumor cerebri. J Pain Symptom Manage 2006; 32(5):401–02.
4 Sinclair AJ, Burdon MA, Nightingale PG et al., Low energy diet and intracranial pressure in women with idiopathic intracranial hypertension: prospective cohort study. BMJ 2010; 340:c2701.
5 Volta U, De Giorgio R, Petrolini N et al., Clinical findings and anti-neuronal antibodies in coeliac disease with neurological disorders. Scand J Gastroenterol 2002; 37:1276–81.
6 Hadjivassiliou M, Davies-Jones GAB, Sanders DS et al., Dietary treatment of gluten ataxia. JNNP 2003; 74(9):1221–24.
7 Hadjivassiliou M, Sanders DS, Grünewald RA et al., Gluten sensitivity: from gut to brain. Lancet Neurol 2010; 9:318–30.

Ursprünglich erschienen:
neuro Supplement 02|2012
neuro Supplement 02|2012

