





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
ÖGN 2012: Diagnostische Entwicklungen: Immunologische Marker
27. April 2012
Biomarker können verwendet werden, um das individuelle Risiko einer Erkrankung abzuschätzen, eine bestehende Erkrankung zu diagnostizieren, den Schweregrad einer Erkrankung zu charakterisieren, einen Therapieeffekt im Organismus zu quantifizieren, die Sicherheit einer Therapie zu beurteilen, PatientInnengruppen zu identifizieren, die nicht von einer Therapie profitieren, und manchmal sogar als therapeutische Ziele dienen.
Die wichtigsten Anforderungen an Biomarker sind der Beweis eines kausalen Zusammenhangs durch Interventionsstudien und/oder experimentelle Modelle, Sensitivität und Spezifität für einen Krankheitsprozess, eine präzise und reproduzierbare labormedizinische Analytik, möglichst leicht zugängliches Probenmaterial und konsistente Effekte in klinischen Studien.
In Tabelle 1 sind wichtige immunologische Biomarker bei neurologischen Erkrankungen zusammengefasst, wobei ich besonders auf immunologische Marker bei demyelinisierenden Erkrankungen eingehen möchte.
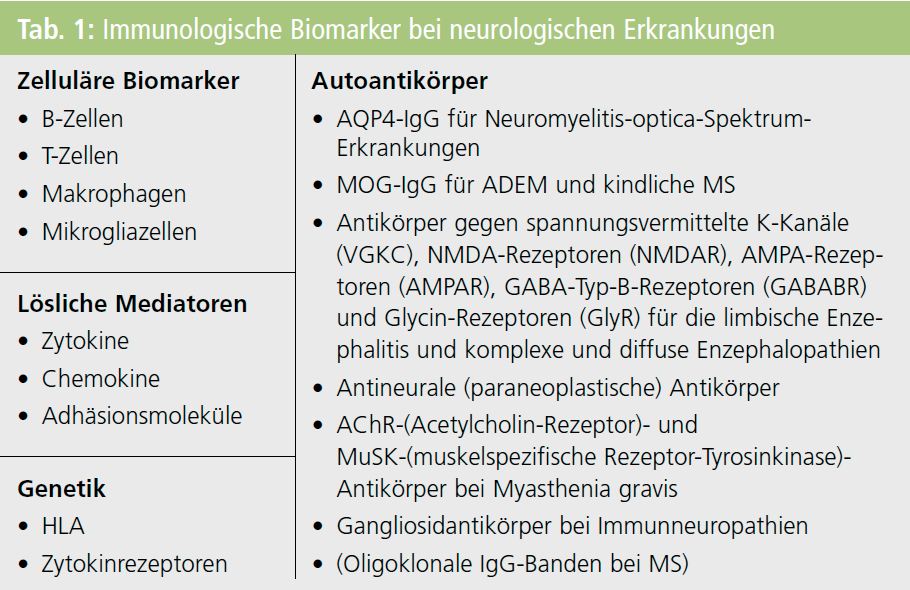
Beispiel AQP4-IgG-Antikörper
Ein wichtiges diagnostisches Problem demyelinisierender Erkrankungen ist die Tatsache, dass sie sich anfänglich meist in Form eines klinisch-isolierten Syndroms (CIS) präsentieren, welches einmalig bleiben oder die Erstmanifestation einer multiplen Sklerose (MS) oder von selteneren demyelinisierenden Erkrankungen wie Neuromyelitis optica (NMO) oder akuter disseminierter Enzephalomyelitis (ADEM) sein kann.
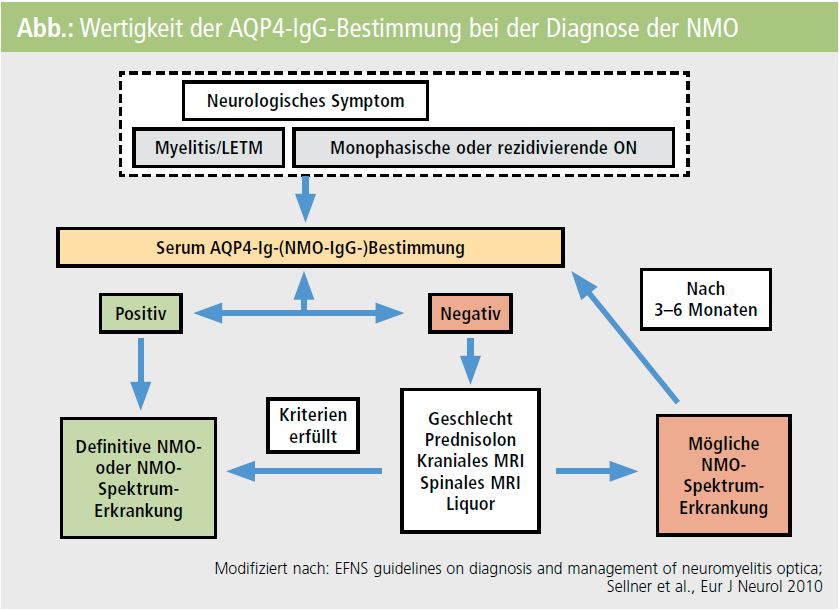
Anhand der NMO lässt sich sehr gut die Entwicklung und Anwendung neuer immunologischer Marker illustrieren, da bei dieser Erkrankung 2004 die sogenannten NMOIgG-Antikörper gegen den Wasserkanal Aquaporin-4 (AQP4) entdeckt wurden2, 3. Die NMO ist eine in unseren Breitengraden eher seltene entzündlich-demyelinisierende Erkrankung des zentralen Nervensystems (Prävalenz 1/105), die klinisch durch das Auftreten einer Optikusneuritis und Myelitis gekennzeichnet ist4. Im Vergleich zur MS haben PatientInnen mit NMO eine schlechtere Prognose und benötigen unterschiedliche Therapien5. Nach der Entdeckung der AQP4-IgGAntikörper als Biomarker der NMO wurden verschiedene Nachweismethoden beschrieben, wobei zellbasierte Testsysteme die höchste Sensitivität und Spezifität aufweisen6–9. Bereits im Jahr 2006 wurden AQP4-IgG (= NMO-IgG) als supportive diagnostische Kriterien der NMO vorgeschlagen10, wobei neben den zwei Leitsymptomen einer Optikusneuritis (ON) und einer Myelitis zwei der drei folgenden Nebenkriterien erfüllt sein müssen:
1) spinales MRT mit langstreckiger Myelonläsion (≥ 3 Wirbelkörpersegmente),
2) ein MS untypisches kraniales MRT bei Erkrankungsbeginn,
3) Nachweis von AQP4-IgG-Antikörper/NMO-IgG-Antikörper im Serum.
Basierend auf diesen diagnostischen Kriterien wurde 2007 die so genannten NMO-Spektrum-Erkrankungen definiert, die nicht nur das Vollbild der NMO, sondern auch AQP4-IgG-positive limitierte Formen wie monophasische oder rezidivierende Myelitiden oder ON enthalten11.
ARGE NMO: In Österreich wurde 2008 auf Initiative von W. Kristoferitsch und F. Aboul-Enein, Neurologische Abteilung SMZ Ost, Wien, M. Storch, Universitätsklinik für Neurologie, Graz, sowie M. Reindl und T. Berger, Universitätsklinik für Neurologie, Innsbruck, die Arbeitsgemeinschaft (ARGE) NMO Österreich etabliert. Im Rahmen der ARGE NMO konnten seit 2008 mit finanzieller Unterstützung der österreichischen MS-Forschungsgesellschaft knapp 2000 Serumproben von über 1500 PatientInnen zahlreicher österreichischer Abteilungen auf AQP4-IgG-Antikörper getestet und dabei 62 PatientInnen (54 Frauen, 56 AQP4-IgG-positiv) mit NMO-Spektrum-Erkrankungen aus 19 Spitälern und Kliniken identifiziert werden. Dies deckt sich sehr gut mit der errechneten Prävalenz der NMO in Österreich, und wir möchten uns bei allen ZuweiserInnen, der ÖGN und der österreichischen MS-Forschungsgesellschaft bedanken.
In der Abbildung ist das diagnostische Vorgehen bei Verdacht auf NMO zusammengefasst.
AQP4-IgG-seronegativ? Trotz der Verbesserung der diagnostischen Tests für AQP4-IgG-Antikörper gibt es eine Subgruppe von NMO-PatientInnen (5–40 %, je nach verwendetem Testsystem), die negativ für AQP4-IgG sind. Es stellt sich daher die Frage, ob diese PatientInnen eine eigene pathogenetische Entität innerhalb der NMO darstellen, oder ob die Seronegativität durch methodische Aspekte (= Sensitivität der AQP4-IgG-Tests) bedingt ist (Tab. 2).
Vor Kurzem konnte gezeigt werden, dass in einer Subgruppe von AQP4-IgG-seronegativen PatientInnen mit NMO, aber auch bei PatientInnen mit Optikusneuritis und/oder Myelitis, Antikörper gegen das Myelin-Oligodendrozyten-Glykoprotein (MOG) vorhanden sind12, 13.
MOG wurde bereits als potenzielles Zielautoantigen der MS in zahlreichen Studien mit kontroversiellen Ergebnissen untersucht, wobei neuere Untersuchungen mit zellbasierten Testsystemen erhöhte MOG-IgG-Titer bei pädiatrischen PatientInnen mit ADEM und MS, nicht aber bei adulter MS zeigten18–21. Die neuen Erkenntnisse über MOG-IgG-Antikörper bei NMO und verwandten Erkrankungen könnten auf eine neue Gruppe von pädiatrischen, aber auch adulten, entmarkenden Erkrankungen mit heterogenen klinischen Manifestationen hindeuten. Allerdings ist MOG sicher nicht das einzige mögliche Zielantigen bei AQP4-IgG-seronegativer NMO. Es ist zu erwarten, dass zukünftige Studien weitere Autoantigene identifizieren werden.

Resümee
Zusammenfassend kann gesagt werden, dass es in den letzten Jahren durch die Etablierung neuer immunologischer Testverfahren gelungen ist, autoantikörpermediierte neurologische Erkrankungen besser zu definieren. Diese eher seltenen, aber oft schwer verlaufenden Erkrankungen, wie die NMO, können so frühzeitig erkannt und richtig behandelt werden. Hingegen konnten bei der „klassischen“ MS trotz intensivster Bemühungen nur wenige Fortschritte bei der Entwicklung neuer spezifischer immunologischer Marker gemacht werden.
1 Biomarkers Definition Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther 2001; 69:89–95.
2 Lennon VA et al., A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004; 364:2106–2112.
3 Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS & Hinson SR, IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. The Journal of Experimental Medicine 2005; 202:473–477.
4 Wingerchuk DM, Hogancamp WF, O‘Brien PC & Weinshenker BG, The clinical course of neuromyelitis optica (Devic‘s syndrome). Neurology 1999; 53:1107–1114.
5 Trebst C et al., Diagnosis and treatment of neuromyelitis optica. Consensus recommendations of the Neuromyelitis Optica Study Group. Nervenarzt 2011; 82:768–777.
6 Takahashi T et al., Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain 2007; 130:1235–1243.
7 Mader S et al., Patterns of antibody binding to aquaporin- 4 isoforms in neuromyelitis optica. PloS one 2010; 5:e10455.
8 Jarius S, Wildemann B, AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nature Rev Neurol 2010; 6:383–392.
9 Waters PJ et al., Serologic diagnosis of NMO: A multicenter comparison of aquaporin-4-IgG assays. Neurology 2012; 78:665–671.
10 Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti, CF, Weinshenker BG, Revised diagnostic criteria for neuromyelitis optica. Neurology 2006; 66:1485–1489.
11 Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG, The spectrum of neuromyelitis optica. Lancet Neurology 2007; 6:805–815.
12 Mader S et al., Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflammation 2011; 8:184.
13 Rostasy K et al., Anti-Myelin Oligodendrocyte Glycoprotein Antibodies in Pediatric Patients With Optic Neuritis. Archives of Neurology 2012; Feb 27. [Epub ahead of print].
14 Kezuka T et al., Relationship Between NMO-Antibody and Anti-MOG Antibody in Optic Neuritis. J Neuroophthalmol 2011; Dec 6. [Epub ahead of print].
15 Ishikawa N, Tajima G, Hyodo S, Takahashi Y, Kobayashi M, Detection of autoantibodies against NMDA-type glutamate receptor in a patient with recurrent optic neuritis and transient cerebral lesions. Neuropediatrics 2007; 38:257–260.
16 Kruer MC et al., NMDA receptor encephalitis mimicking seronegative neuromyelitis optica. Neurology 2010; 74:1473–1475.
17 Jarius S, Wandinger KP, Borowski K, Stoecker W, Wildemann B, Antibodies to CV2/CRMP5 in neuromyelitis optica-like disease: Case report and review of the literature. Clin Neurol Neurosurg 2011; [Epub ahead of print].
18 Di Pauli F et al., Temporal dynamics of anti-MOG antibodies in CNS demyelinating diseases. Clin Immunol 2011; 138:247–254.
19 O‘Connor KC et al., Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nature medicine 2007; 13:211–217.
20 Brilot F et al., Antibodies to native myelin oligodendrocyte glycoprotein in children with inflammatory demyelinating central nervous system disease. Ann Neurol 2009; 66:833–842.
21 Lalive P et al., Highly reactive anti-myelin oligodendrocyte glycoprotein antibodies differentiate demyelinating diseases from viral encephalitis in children. Mult Scler 2011; 17:297–302.

Ursprünglich erschienen:
neuro Supplement 02|2012
neuro Supplement 02|2012

