





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Ichthyosis vulgaris – Filaggrin-Genotyp beeinflusst epidermale Barrierestörung
25. November 2011
Seit der Entdeckung von Mutationen im Filaggrin-Gen als molekulare Ursache für die Ichthyosis vulgaris im Jahr 2006 sind durch die Ergebnisse zahlreicher internationaler Studien die wichtige Rolle von Filaggrin für die Hautbarrierefunktion und der Barrieredefekt bei Patienten mit Ichthyosis vulgaris und atopischer Dermatitis hinreichend gesichert. Nach wie vor ungeklärt ist jedoch der Pathomechanismus, auf welche Weise der Mangel an Filaggrin die Schwächung der Hautbarriere hervorruft.
Das Akronym Filaggrin leitet sich vom englischen „filament aggregating protein“ ab. Diese Benennung wurde gewählt, weil im Rahmen der Differenzierung der Keratinozyten Filaggrin die Aggregation von Keratinfilamenten im Stratum granulosum fördert. Die einzelnen Filaggrin-Monomere entstehen durch Dephosphorylierung und proteolytischen Zerfall aus dem Pro-Filaggrin, das im oberen Stratum spinosum synthetisiert wird und im Stratum granulosum den Hauptbestandteil der Keratohyalingranula ausmacht. Im Stratum corneum sind die Filaggrin-Abbauprodukte (hygroskopische Aminosäuren und Urocansäure) für die natürliche Rückfeuchtung und den sauren pH-Wert der Haut verantwortlich. In gesunder Hornschicht wird die Hautbarrierefunktion vorwiegend durch die extrazellulären Lipide aufrechterhalten. Corneodesmosomen und Tight Junctions spielen ebenfalls eine wichtige, aber noch besser zu definierende Rolle. Wie Filaggrin als interzelluläres Strukturprotein diese extrazellulären Strukturen reguliert, ist jedoch auf den ersten Blick nicht offensichtlich.
Studie: Die Rolle von Filaggrin
In einer kürzlich im American Journal of Pathology publizierten Arbeit haben wir gemeinsam mit Kooperationspartnern aus Wien, Hamburg, Dundee, Brüssel, Seattle und San Francisco diese Frage näher beleuchtet und neue, interessante Erkenntnisse zur epidermalen Struktur und Funktion in Filaggrin-defizienter Haut gefunden.
Um zu klären, wodurch im Falle einer Mutation im Filaggrin-Gen und in deren Folge durch einen epidermalen Filaggrin-Mangel der Hautbarrieredefekt verursacht wird, wurden im Rahmen der Arbeit 19 Patienten mit Ichthyosis vulgaris und Filaggrin-Mutationen sowie 20 gematchte Kontrollpersonen ohne Defekt im Filaggrin-Gen miteinander auf funktioneller und ultrastruktureller Ebene verglichen.
Ergebnisse
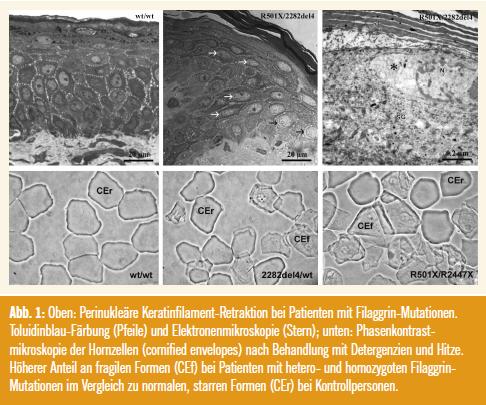
Histologisch und in der immunhistochemischen Färbung auf Filaggrin wiesen homozygote (compound-heterozygote) Ichthyose-Patienten einen kompletten Verlust und heterozygote eine Reduktion des Stratum granulosum auf. Die Keratinaggregation zeigte elektronenmikroskopisch in Patienten mit Filaggrin-Mutationen perinukleäre Unregelmäßigkeiten – perinukleäre Halos – (Abb. 1), der Hautoberflächen-pH-Wert war erhöht und die Hydratation des Stratum corneum erniedrigt. Überraschenderweise waren die aus Hautschuppen durch Kochen in einer speziellen Pufferlösung isolierten Hornzellen (sog. cornified envelopes) auch bei den Ichthyose-Patienten weitgehend intakt. Lediglich bei Exposition gegenüber starken Detergenzien und Hitze sowie mechanischem Stress (Ultraschall) waren die Filaggrin-defizienten Korneozyten fragiler (Abb. 1). Hingegen war die Organisation der extrazellulären Lipidlamellen im Stratum corneum von Patienten mit Filaggrin-Mutationen in der Elektronenmikroskopie deutlich verändert, neben Diskontinuitäten in den Lamellen war eine verzögerte Ausschleusung des Lipidmaterials aus den Korneozyten in den Interzellularraum nachweisbar. Zusätzlich waren die Zell-Zell-Kontakte der Hornzellen auffällig, weil die Zahl der Corneodesmosomen und Tight Junctions bei den Ichthyose-Patienten deutlich reduziert war. Eine mögliche Erklärung für diese Beobachtungen ist, dass der erhöhte pH-Wert im Stratum corneum von Filaggrin-defizienter Haut die Aktivität einiger wichtiger, streng pH-Wertabhängiger Enzyme (Proteasen, Lipidenzyme) beinträchtigen könnte.
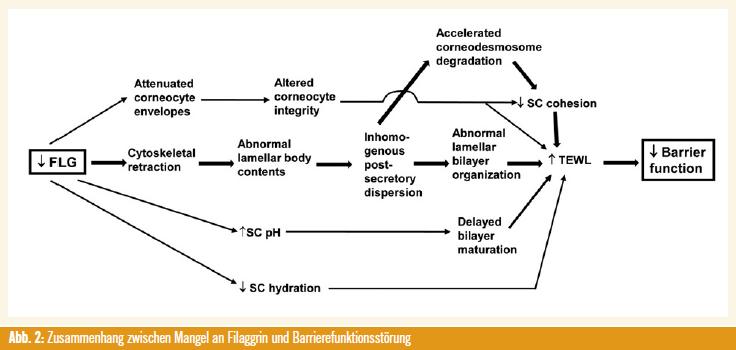
Zusammenfassend konnte in der vorliegenden Arbeit gezeigt werden, dass durch einen Mangel an Filaggrin Veränderungen in der Barrierearchitektur hervorgerufen werden (Abb. 2).

Ursprünglich erschienen:
SD 02|2011
SD 02|2011
Herausgeber: Univ.-Prof. Dr. Hubert Pehamberger; Ao. Univ.-Prof. Dr. Rainer Kunstfeld
Publikationsdatum: 2011-11-25
Zur Ausgabe »
Publikationsdatum: 2011-11-25
Zur Ausgabe »
