
























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Interaktionen von Opioiden mit nichtopioiden Analgetika und Koanalgetika
1. März 2013
Wechselwirkungen zwischen Arzneimitteln („drug-drug interactions“) sind häufig Ursache für unerwünschte Arzneimittelwirkungen (UAW). Risikofaktoren wie Multimorbidität, höheres Alter sowie Polypharmazie führen zu vermehrtem Auftreten von UAW, die Grund für eine Krankenhausaufnahme oder einen verlängerten Krankenhausaufenthalt sein können. Klinisch relevante pharmakokinetische und -dynamische Interaktionen treten auch zwischen den in der Analgesie und Substitutionstherapie eingesetzten Opioiden und nichtopioiden Analgetika sowie Koanalgetika (Antidepressiva, Antikonvulsiva) auf.
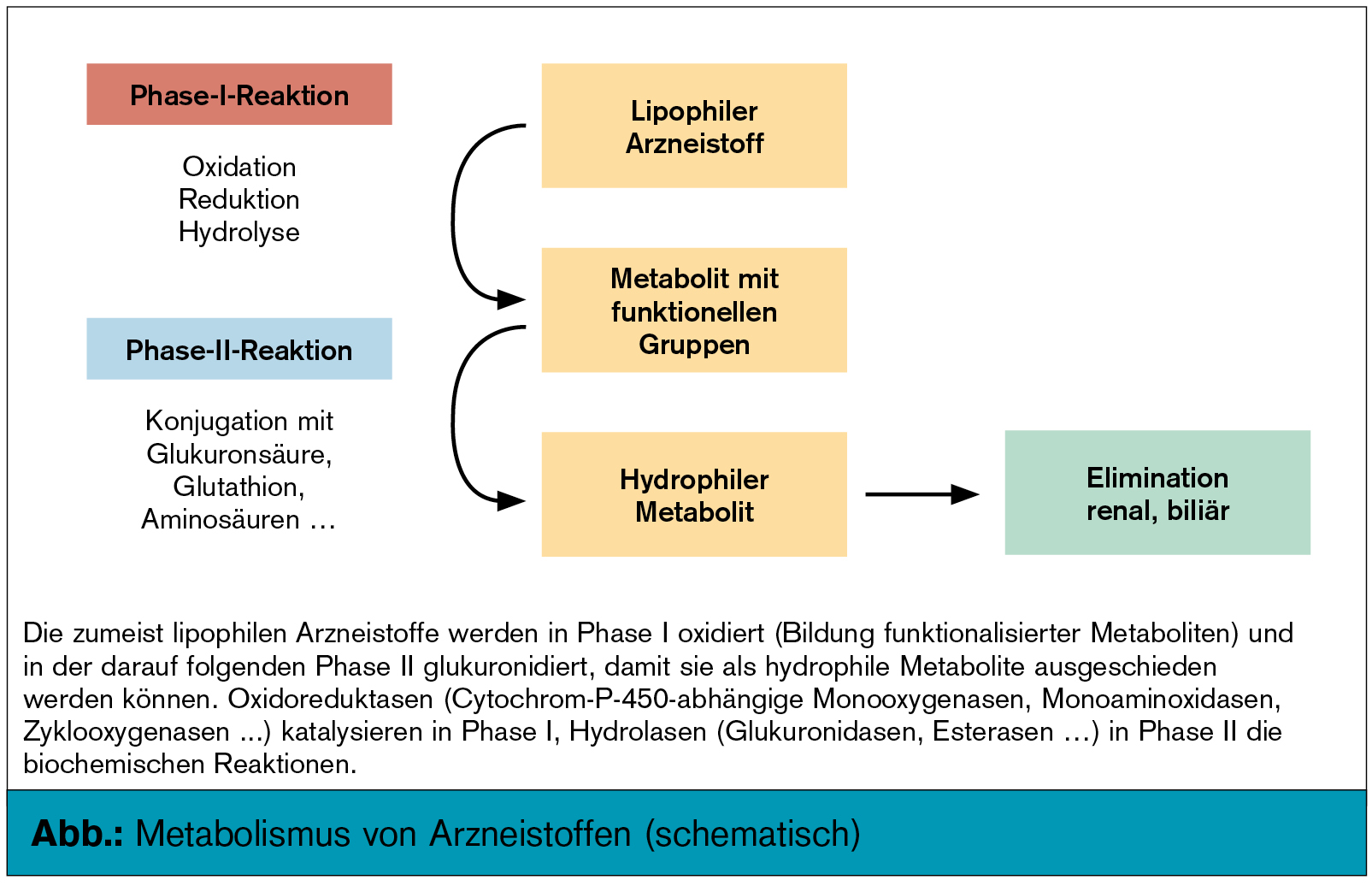
Die veränderte therapeutische Wirkung, bedingt durch pharmakokinetische Interaktionen, ist mit Konzentrationsänderungen der gleichzeitig verabreichten Arzneistoffe verbunden. Alter, Geschlecht, genetischer Polymorphismus und Organinsuffizienzen beeinflussen diesen Prozess wesentlich. Dem gegenüber stehen pharmakodynamische Interaktionen, bei denen die veränderte Wirkung am Wirkort im Sinne eines Synergismus oder Antagonismus ohne Änderung der Konzentrations-Zeit-Profile zustande kommt. Das für pharmakokinetische „drug-drug interactions“ wichtigste Enzymsystem ist jenes der Zytochrom-P450-abhängigen Monooxygenasen (CYP450). Deren Aktivität und damit der Arzneistoffmetabolismus (Abb.) kann durch andere Arzneistoffe und Lebensmittel erhöht (Enzyminduktion) oder vermindert (Enzyminhibition) werden. Für den Metabolismus der Opioide sind insbesondere die Isoformen CYP3A4 und CYP2D6 von Bedeutung.
Verstärkung der serotoninagonistischen Wirkung: Bei Komedikation der proserotonergen Opioide Methadon und Tramadol als Prodrug kommt es aufgrund der eingeschränkten Bildung des aktiven Hauptmetaboliten O-Desmethyltramadol einerseits zu einer verringerten analgetischen Wirkung und andererseits zu verstärkten serotonergen Nebenwirkungen.
Es ist daher auf Symptome einer serotonergen Überstimulation wie exzessives Schwitzen, Hypertonie, motorische Unruhe oder Hyperreflexie zu achten. Über den gleichen Mechanismus – die Blockierung des Tramadolabbaus über eingeschränkte CYP2D6-Enzymaktivität – läuft die Interaktion mit dem Setrotonin-Noradrenalin-Wiederaufnahmehemmer (SNRI) Duloxetin. Auch andere CYP2D6-Inhibitoren, die in der Schmerztherapie nicht primär verwendet werden wie Paroxetin oder Sertralin verursachen diese pharmakokinetische Interaktion. Das ebenfalls als Prodrug vorliegende Dihydrokodein unterliegt denselben pharmakokinetischen Wechselwirkungen. Additive serotonerge Effekte können sich als UAW auch mit Fentanyl, Pethidin oder Dextromethorphan (in rezeptfreien Antitussiva enthalten) und dem proserotonergen trizyklischen Antidepressivum Amitriptylin sowie dem SNRI Venlafaxin ergeben. Besonders ältere Menschen reagieren häufig mit verstärkter Unruhe und Halluzinationen auf diese pharmakodynamische Interaktion. Hydromorphon eignet sich bei starken Schmerzen aufgrund des sehr geringen Interaktionspotenzials besser für diese spezielle Patientengruppe.
Hyponatriämie unter Opioiden, NSAR, Antidepressiva und Antikonvulsiva: Eine andere Ursache für Verwirrtheitszustände, Übelkeit und erhöhte Sturzgefahr – besonders bei Älteren – stellt das Syndrom der inadäquaten ADH-Sekretion (SIADH) dar. Dabei kommt es zu einer gemäß der Serumosmolarität inadäquat hohen Freisetzung von Adiuretin. Dadurch wird vermehrt freies Wasser in der Niere rückresorbiert, was zu einer Verdünnungshyponatriämie mit einem Absinken des Serumnatriums (hypoosmolare Hyponatriämie) auf Werte < 135 mmol/l führt. Die Therapie mit Morphin, Tramadol, trizyklischen Antidepressiva, Duloxetin, Venlafaxin oder Carbamazepin sowie mit nichtsteroidalen Antirheumatika per se oder in Kombination zählt zu den medikamentösen Ursachen für diese UAW. Je nach Schwere der neurologischen Symptomatik liegt die Therapie in der Restriktion der Flüssigkeitszufuhr (Cave! Überwässerung), der vorsichtigen Substitution von NaCl (Essen salzen, hypertone NaCl-Lösung) und eventueller Gabe von Furosemid.
Bedeutung der UDP-Glukuronosyltransferasen für den Metabolismus von Morphin: Die UDP-Glukuronosyltransferasen (UGT) werden zunehmend bezüglich ihrer Bedeutung für Arzneimittelinteraktionen erkannt. Lokalisiert an der luminalen Seite des endoplasmatischen Retikulums katalysieren sie die Umwandlung lipophiler in hydrophile, renal ausscheidbare Substanzen. Morphin wird im Vergleich zu den anderen Opioiden minimal über das CYP450-Enzymsystem metabolisiert, sondern hauptsächlich über die UDP-Glukuronosyltransferase UGT2B7 zu Morphin-3-glukuronid (kein analgetischer Effekt) und Morphin-6-glukuronid (analgetisch potenter wirksam als Morphin) abgebaut. In-vitro-Untersuchungen ergaben, dass die Komedikation von Morphin mit Inhibitoren von UGT2B7 wie den Koanalgetika Amitriptylin, Diclofenac, Naproxen und Carbamazepin aber auch mit den Benzodiazepinen Oxazepam und Diazepam die Glukuronidierung blockiert und zu verstärkter Morphinwirkung führt, was einen additiven analgetischen Effekt, aber auch eine erhöhte Nebenwirkungsrate mit verstärkter Atemdepression und Sedierung – besonders in der Kombination mit Benzodiazepinen – nach sich ziehen kann.
Erhöhtes Risiko für Herzrhythmusstörungen durch Verlängerung des QT-Intervalls: Ursache für eine medikamenteninduzierte Verlängerung des QT-Intervalls auf zellulärer Ebene ist die Hemmung des transmembranären Kaliumstroms, der maßgeblich für die Rückkehr des Aktionspotenzials zum Ruhemembranpotenzial verantwortlich ist. Im Oberflächen-EKG zeigt sich dieser Effekt in der Verlängerung des QT-Intervalls um 45–60 ms. Wird das Membranpotenzial aufgrund einer abnormen QT-Intervall-Verlängerung instabil, können als Folge so genannte frühe Nachdepolarisationen auftreten. Erreichen diese die Schwelle für die Auslösung eines neuen Aktionspotenzials, kommt es zur Ausbildung einer getriggerten Aktivität, den Torsades de pointes (TdP). TdP enden meist spontan mit Schwindel und Synkopen, sie können aber auch in Kammerflimmern und damit Letalität münden.
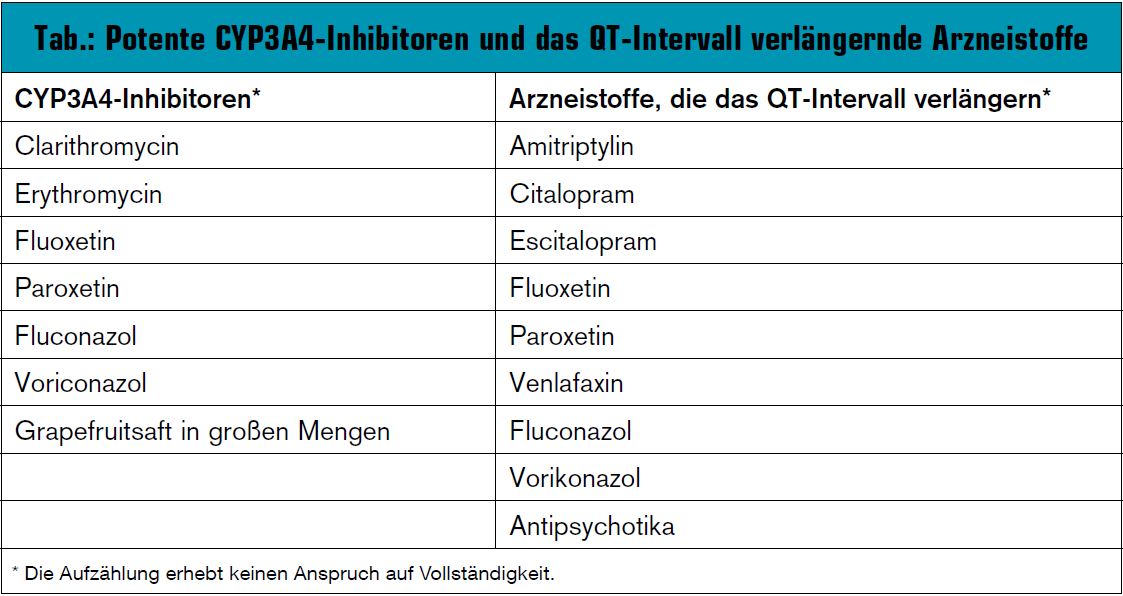
Da die Dauer des QT-Intervalls von der Herzfrequenz abhängig ist, wird als Vergleich die frequenzkorrigierte QT-Zeit herangezogen (QTc). Die Therapie mit Methadon und in geringerem Ausmaß mit Levomethadon weist besonders in Kombination mit Arzneistoffen, die deren Abbau hemmen (v. a. CYP3A4-Inhibitoren) und/oder Substanzen, die ebenfalls das QT-Intervall verlängern, ein erhöhtes Risiko für diese kardiale Nebenwirkung auf (Tab.).
Neben Methadon wird auch Buprenorphin über das Isoenzym CYP3A4 umgesetzt, das per se das QT-Intervall aber nicht verlängert. Mit obigen, beispielhaft genannten CYP3A4-Inhibitoren können sich erhöhte Plasmaspiegel von Buprenorphin und damit verstärkt opioidtypische Nebenwirkungen ergeben.
Abfall der Plasmaspiegel und Wirkverlust durch Carbamazepin: Carbamazepin kann als starker Induktor von CYP3A und CYP2B6 in Komedikation mit einigen Opioiden und den als Koanalgetika eingesetzten Antidepressiva deren Abbau induzieren, was zu verringerten Plasmaspiegeln und damit zu Wirkverlust führt: Methadon, Fentanyl, Pethidin, Buprenorphin, Dihydrokodein, Tramadol sowie Amitriptylin und Duloxetin interagieren mit Carbamazepin. Zu beachten ist der damit verbundene Latenzeffekt: Die Induktion des Abbaus setzt bei Zusatz von Carbamazepin zu einer bereits bestehenden Therapie mit einer zeitlichen Verzögerung von 7–10 Tagen voll ein, dessen Aufhebung bei Absetzen des Antikonvulsivums dauert ebenso lang. Die Kombination Carbamazepin und Buprenorphin wird in der Entzugstherapie opioidabhängiger Patienten eingesetzt. Der induktive Effekt von Carbamazepin ist jedoch in der Schmerztherapie unerwünscht.
Nicht sinnvoll: Abschließend soll auf nicht sinnvolle Kombinationen eingegangen werden. Die Kombination von Buprenorphin mit reinen Opioidagonisten wie Morphin, Methadon, Fentanyl, Oxycodon, Piritramid oder Dihydrokodein haben im klinisch empfohlenen Dosisbereich keine Relevanz.
Bei Patienten, die bereits eine gewisse Opiattoleranz entwickelt haben, ist in dieser Kombination mit einem Entzugssyndrom zu rechnen. Generell wird gemäß WHO-Stufenschema zur Schmerztherapie eine Kombination von Opioiden verschiedener Wirkstärken nicht empfohlen.
Faustregel: Auch unter Zuhilfenahme diverser Interaktionstools und Beachtung der individuellen Patientenparameter ist es oft nicht einfach, die klinische Relevanz von Arzneimittelinteraktionen und der sich daraus ergebenden, unerwünschten Arzneimittelwirkungen zu beurteilen. Es gilt die mehrfach zitierte Faustregel: Eine geringe therapeutische Breite des Arzneistoffs und eine steile Dosis-Wirkung-Kurve, bestehende Organinsuffizienzen und das meist damit verbundene höhere Alter des Patienten steigern das Risiko für klinisch relevante Arzneimittelinteraktionen.
Literatur bei der Verfasserin

AutorIn: Mag. pharm. Sonja Mayer, AHPH
Apotheke, Landes-Nervenklinik Wagner-Jauregg, Linz

Ursprünglich erschienen:
UIM 01|2013
UIM 01|2013