





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Die kardiale Transthyretin-(ATTR-)-Amyloidose ist eine infiltrative Herzmuskelerkrankung, die als vererbbare (ATTRv-Amyloidose) oder nichtvererbbare Form (ATTRwt-Amyloidose) auftreten kann. Ursächlich ist der Zerfall von ATTR-Tetrameren aus der Leber in Monomere, die sich als unlösliche Amyloidfibrillen im Extrazellulärraum des Myokards ablagern und zu einer Verdickung des Herzmuskels führen. Begleitend können auch extrakardiale Amyloid-Ablagerungen vorkommen. Die kardiale ATTR-Amyloidose blieb in der Vergangenheit oft unerkannt. Fortschritte in der Diagnostik und Behandlung haben in den letzten Jahren das Bewusstsein für diese Erkrankung erhöht.
Klinik
Es gibt zahlreiche kardiale und extrakardiale Red Flags, die auf eine zugrunde liegende kardiale ATTR-Amyloidose hinweisen können. Die klinische Symptomatik variiert je nach betroffenen Organen und Ausmaß der Organbeteiligung. Eine kardiale Beteiligung kann sich mitunter unspezifisch als Herzinsuffizienz mit Schwäche, Dyspnoe und peripheren Stauungszeichen manifestieren. Aber auch bradykarde oder tachykarde Herzrhythmusstörungen mit Schwindel, Synkopen bis hin zum plötzlichen Herztod können mit einer Amyloidose assoziiert sein. Es ist zu beachten, dass extrakardiale Amyloidose-Manifestationen einer kardialen Erstdiagnose um Jahre vorausgehen können. Besonders muskuloskelettale Beschwerden sollten an eine ATTR-Amyloidose denken lassen (z.B. Karpaltunnelsyndrom, Spinalkanalstenose, Bizepssehnenruptur).
Diagnose
Bei klinischem Verdacht auf Amyloidose kann die Echokardiografie entscheidende Hinweise für den Erkrankungsnachweis liefern. Im Frühstadium zeigt sich meist eine diastolische Dysfunktion, während im fortgeschrittenen Stadium auch die systolische Funktion beeinträchtigt sein kann. Weitere charakteristische Merkmale sind eine biventrikuläre Hypertrophie, eine biatriale Dilatation, verdickte Klappen und ein hämodynamisch nichtwirksamer Perikarderguss. Typisch ist auch das Apical-Sparing-Muster in der Strain-Analyse (=reduzierter basaler longitudinaler Strain bei erhaltenem apikalem Strain). Zur Abgrenzung von anderen kardialen Hypertrophie-Erkrankungen stellt das Herz-MRT eine wertvolle Bildgebungsmethode dar. Diese erlaubt neben dem Erkennen struktureller/funktioneller Abnormalitäten eine detaillierte Myokard-Charakterisierung (typisches Late-Enhancement-Muster, erhöhte T1-Relaxationszeit, erhöhtes extrazelluläres Volumen hinweisend für kardiales Amyloid) (Abb. 1).
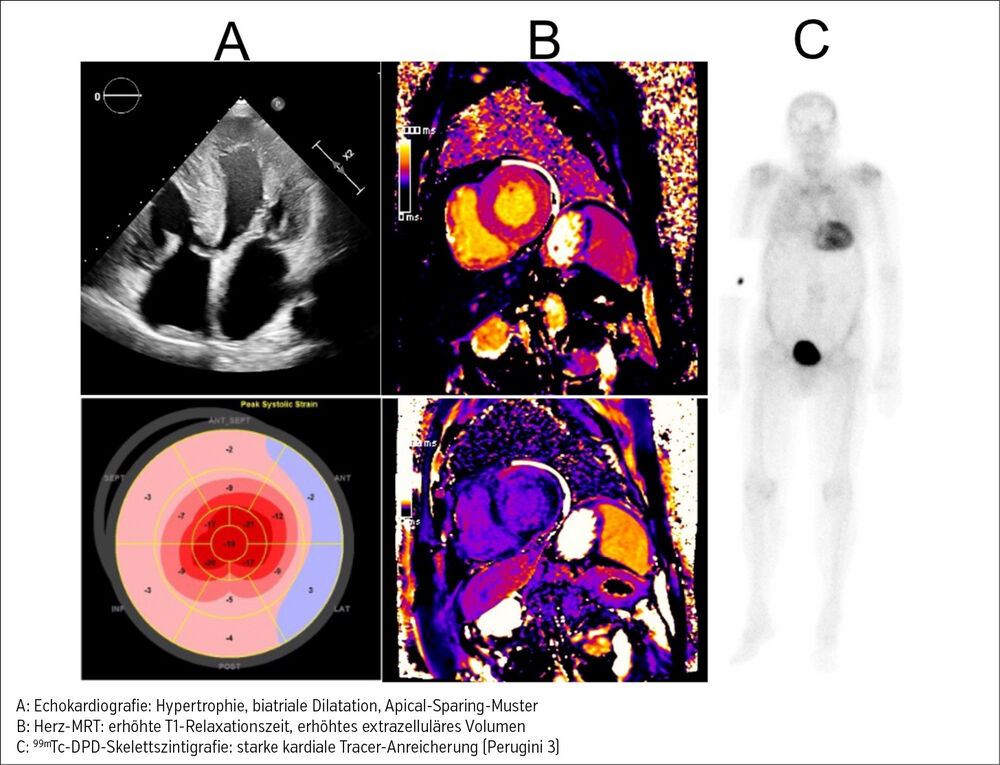
Abb. 1: Amyloidose-Merkmale in der Bildgebung
Weder Echokardiografie noch Herz-MRT sind jedoch allein ausreichend für eine endgültige Diagnosestellung. Die Diagnose kann heutzutage dennoch mehrheitlich nichtinvasiv gesichert werden anhand einer negativen Leichtkettendiagnostik und einer positiven Skelettszintigrafie. Erstere umfasst die Bestimmung der freien Leichtketten, eine Serum-/Harneiweißelektrophorese mit Immunfixation und dient dem Ausschluss einer Leichtketten-Amyloidose. Dieser liegt eine Plasmazellerkrankung zugrunde und sollte frühzeitig detektiert werden, da diese unbehandelt mit einer schlechten Prognose verbunden ist. Die Skelettszintigrafie erfordert die Applikation eines radioaktiven Tracers (99mTc-DPD, 99mTc-PYP, 99mTc-HMDP), woraufhin das Ausmaß der kardialen Tracer-Anreicherung (planar, SPECT/CT) nach Perugini von 0 (=keine Anreicherung) bis 3 (=deutliche Anreicherung) graduiert wird (Abb. 1).
Die Kombination aus negativer Leichtkettendiagnostik und starker Tracer-Anreicherung in der Skelettszintigrafie (=Perugini ≥2) ergibt die Diagnose einer kardialen ATTR-Amyloidose. Im Anschluss wird unabhängig vom Alter der Patient:innen eine genetische Testung empfohlen (Abb. 2).
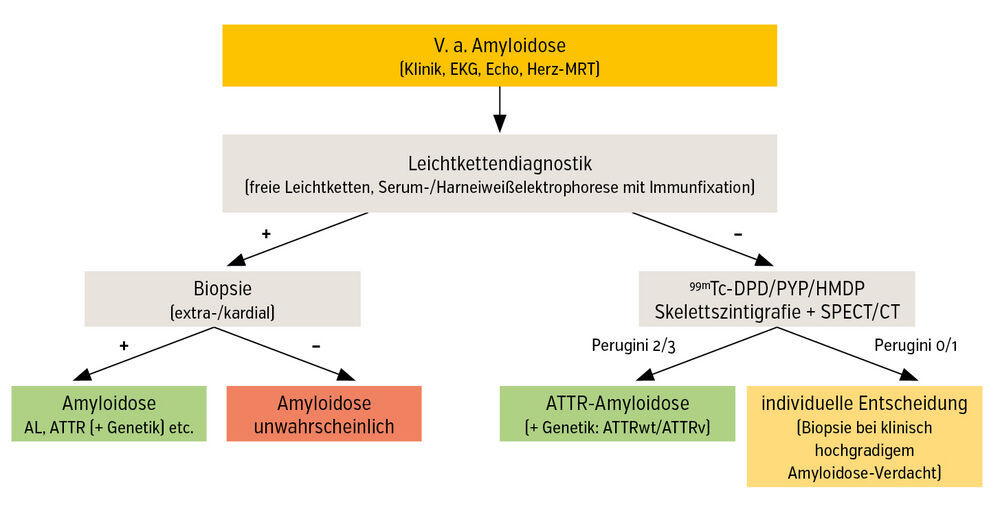
Abb. 2: Diagnosealgorithmus der kardialen Amyloidose
Sind die nichtinvasiven Diagnosekriterien nicht erfüllt (=positive Leichtkettendiagnostik und/oder Perugini <2 bei klinisch begründetem Amyloidose-Verdacht), ist eine Biopsie indiziert (Abb. 2). Je nach Zentrumserfahrung und Ausmaß der Organbeteiligung kann entweder eine Herzmuskelbiopsie oder eine extrakardiale Biopsie inklusive Amyloid-Typisierung durchgeführt werden.
Therapie
Zur symptomorientierten Behandlung der kardialen ATTR-Amyloidose werden Schleifendiuretika eingesetzt, wobei eine Euvolämie angestrebt wird. Aldosteronantagonisten und SGLT2-Inhibitoren ergänzen die Therapie und werden meist gut toleriert. Vorsicht ist geboten bei der Anwendung von RAAS-Blockern und Beta-Blockern. Diese werden aufgrund von Hypotonieneigung und chronotroper Inkompetenz oft schlecht vertragen. Aufgrund des hohen Thromboembolierisikos wird bei ATTR-Patient:innen mit Vorhofflimmern unabhängig vom CHA2DS2-VA-Score eine Empfehlung zur oralen Antikoagulation ausgesprochen.
Krankheitsspezifische ATTR-Therapien umfassen Stabilisatoren und Gene Silencer, die direkt in die Amyloid-Pathogenese eingreifen. Tafamidis erlaubt eine ATTR-Tetramerstabilisierung und ist bislang das einzige zugelassene Medikament für die kardiale ATTRwt-Amyloidose. Die Gene Silencer Patisiran, Inotersen und Vutrisiran werden bereits bei der ATTRv-Amyloidose mit Polyneuropathie eingesetzt. Die Wirksamkeit und Sicherheit von Vutrisiran konnte kürzlich auch für das kardiale ATTR-Kollektiv (ATTRwt, ATTRv) gezeigt werden.
Besonderes Interesse gilt derzeit einem monoklonalen Antikörper (NI006) und einem CRISPR/Cas9-basierten Therapieansatz (NTLA-2001), die aktuell bei ATTR-Patient:innen untersucht werden. Vor allem die antikörperbasierte Therapie gibt Hoffnung auf eine Wiederherstellung der strukturellen und funktionellen Organfunktionen.

Autorin:
Dr.in Maria Ungericht
Dr.in Maria Ungericht
Universitätsklinik für Innere Medizin III – Kardiologie und Angiologie, Medizinische Universität Innsbruck

Autor:
Assoc. Prof. Priv.-Doz. Dr. Marc-Michael Zaruba
Assoc. Prof. Priv.-Doz. Dr. Marc-Michael Zaruba
Klinik für Innere Medizin III, Kardiologie und Angiologie, Interdisziplinäres Herzinsuffizienzzentrum Tirol, IHZ, Expertisezentrum für seltene Herzmuskelerkrankungen, Medizinische Universität Innsbruck

Ursprünglich erschienen:
AEK 22|2024
AEK 22|2024
WAS PATIENT:INNEN WISSEN WOLLEN
- Schwindel und/oder Synkopen sind als Warnsignale zu werten. Eine Suche nach Brady-/Arrhythmien mittels Langzeit-EKG ist ratsam und soll jährlich wiederholt werden.
- Bei Nachweis einer ATTRv-Amyloidose soll ein Familienscreening durchgeführt werden. ATTRv-Patient:innen sollten an ein Amyloidose-Zentrum angebunden werden.
- Eine krankheitsspezifische Therapie soll möglichst früh im Krankheitsverlauf begonnen werden, um weitere Amyloid-Ablagerungen zu verhindern.
- Tafamidis ist das bisher einzige zugelassene Medikament zur Behandlung der kardialen ATTRwt-Amyloidose und wird gut vertragen, ohne Notwendigkeit spezieller Laborkontrollen. Die Therapie muss alle 6 Monate über ein Amyloidose-Zentrum erneut beantragt werden.
- Klinik (NYHA-Klasse, Diuretika-Bedarf), Laborparameter (eGFR, NT-proBNP, Troponin T) und Bildgebung erlauben eine Risikostratifizierung (Therapie-Monitoring).
WAS PATIENT:INNEN WISSEN WOLLEN
Kann die Erkrankung geheilt werden?
Nein, die derzeit zugelassenen Medikamente ermöglichen eine Stabilisierung der Erkrankung, jedoch keine Heilung. Eine lebenslange Therapiefortführung ist bislang erforderlich.
Können Kribbelparästhesien ein Anzeichen für eine ATTR-Amyloidose sein?
Ja, solche Beschwerden können auf eine neurologische Beteiligung im Rahmen einer ATTR-Amyloidose hinweisen. In diesem Fall sollte eine neurologische Abklärung erfolgen.
Literatur bei den Verfasser:innen