





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Früherkennung der Lungenfibrose
22. September 2017
Die idiopathische pulmonale Fibrose (IPF) ist eine seltene Erkrankung. Sie ist unter den idiopathischen interstitiellen Pneumonien die relativ häufigste Form, die Ursachen sind ungeklärt. Hinsichtlich der Pathogenese wird derzeit angenommen, dass es durch apoptotische Alveolarepithelzellen zu folgenden Prozessen kommt:
(1) gestörte Regeneration, (2) Aktivierung von Fibroblasten, (3) Differenzierung zu Myofibroblasten mit verstärkter Proliferation und (4) Ablagerung von extrazellulärer Matrix. Die Lunge vernarbt zunehmend, wobei gesundes Lungengewebe in Form der Lungenbläschen und der Blutkapillaren verloren geht. Der Körper kann nicht mehr ausreichend mit Sauerstoff versorgt werden.1
Epidemiologie
Die Erkrankung betrifft hauptsächlich Erwachsene – Männer häufiger als Frauen – ab dem 50. Lebensjahr. Zudem gibt es eine starke Assoziation zwischen Rauchen und IPF. Die Prävalenz liegt bei zwei bis 29 pro 100.000 innerhalb der Allgemeinbevölkerung – damit ist die IPF per Definition eine seltene Erkrankung (Orphan Disease), allerdings ist die Prävalenz in der Altersgruppe der Über-70-Jährigen um bis zu 10-fach höher.1
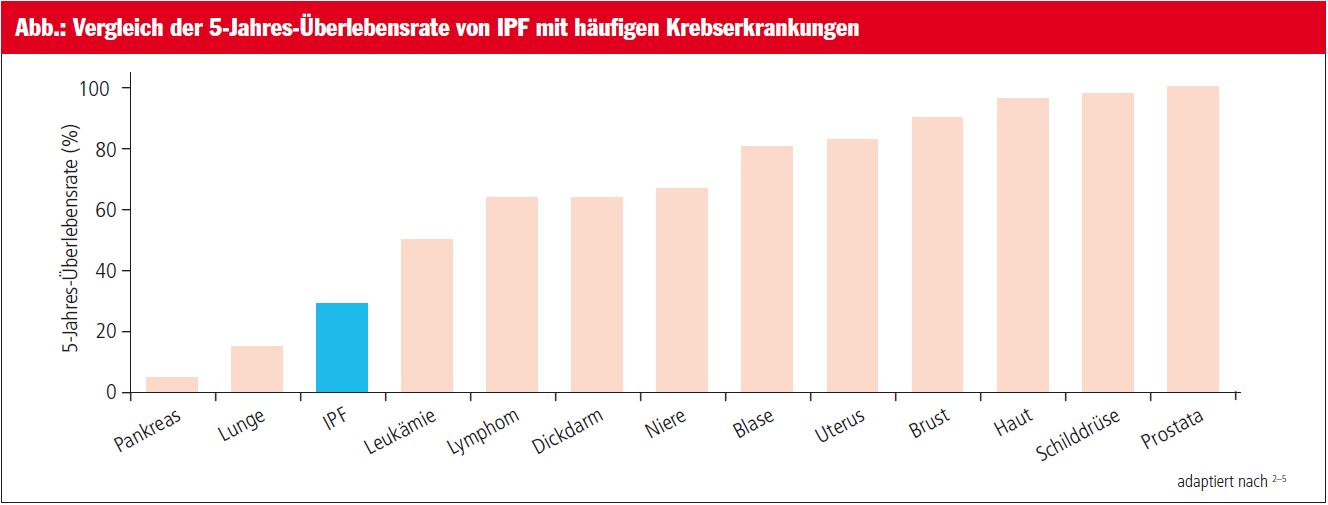
Die Prognose der IPF ist ungünstiger als die vieler Krebserkrankungen (Abb.).1–5 Die mediane Überlebenszeit beträgt 3–4 Jahre nach Diagnosestellung, bei rasch progredient verlaufender IPF verkürzt sich die mediane Lebenserwartung auf weniger als 2 Jahre.1, 6
Diagnose und Therapie
Das wichtigste radiologische Verfahren für die Diagnose der IPF ist die High-Resolution-Computertomografie (HRCT). Typische Kriterien des UIP-(„usual interstitial pneumonia“-)Musters sind retikuläre Verdichtungen, eine Zunahme der fibrotischen Veränderungen von apikal nach basal mit Betonung der Lungenperipherie sowie ein Honigwabenmuster, das oft zusammen mit Traktionsbronchiektasen (= erweiterte Bronchien durch Schrumpfung des umliegenden Lungengewebes) auftritt. Bei Vorliegen von extensiven Milchglasarealen, von Konsolidierungen und Zysten sowie von Granulomen und Mikrogranulomen kann eine IPF dagegen ausgeschlossen werden. Wenn die HRCT kein sicheres UIP-Muster zeigt, ist eine chirurgische Lungenbiopsie indiziert.
Medikamente könnten die Progression der IPF verzögern, aber wahrscheinlich nicht dauerhaft stoppen. Aktuell stehen zur Behandlung der IPF zwei verschiedene antifibrotisch wirksame Medikamente zur Verfügung: Pirfenidon und Nintedanib.7 Während vor der Zulassung dieser Antifibrotika nur eine symptomatische Therapie der IPF möglich war, kann der Verlauf der Erkrankung durch eine Therapie mit Pirfenidon oder Nintedanib nachweislich verlangsamt werden: je früher der Therapiebeginn, desto positiver die Auswirkung auf den Krankheitsverlauf.
CT-Screening zur Früherkennung
Ein regelmäßiges Screening mit niedrigdosierter Computertomografie (CT) konnte die lungenkrebsspezifische Mortalität bei asymptomatischen Personen mit hohem Risiko für Lungenkrebs signifikant senken, so das Ergebnis des National Lung Screening Trial (NLST).8
Die CT-Bilder einer Screening-Untersuchung können neben dem Nachweis von Vorstufen von Lungenkarzinomen (sogenannte Rundherden) auch zur Früherkennung weiterer Erkrankungen wie zum Beispiel Osteoporose, Erkrankungen der Koronararterien oder IPF genutzt werden. Auf diese Weise ließe sich die Effizienz eines Screening-Programms weiter verbessern. Daher wäre es durchaus sinnvoll, im Rahmen eines Lungenkarzinom-Screenings nicht nur auf auffällige Rundherde zu achten, sondern auch andere wichtige rauchassoziierte Erkrankungen im Frühstadium zu diagnostizieren. Eine retrospektive Analyse der NLST-Studie fand bei einem nicht unerheblichen Teil der Probanden (55–74 Jahre alt, schwere Raucher bzw. Ex-Raucher) Veränderungen, die auf eine Lungenfibrose hinweisen (interstitielle Abnormalitäten). Bei 9,7 % der Studienteilnehmer zeigten sich teilweise fibrotische, aber auch nichtfibrotische Veränderungen. Bei 37 % der Patienten mit fibrotischen Veränderungen kam es zu einem Fortschreiten der Erkrankung.9
Dass eine Früherkennung von solchen interstitiellen Abnormalitäten relevant sein kann, zeigte eine Metaanalayse mehrerer großer COPD-Studien. In dieser Analyse konnte gezeigt werden, dass bei Patienten mit COPD das Vorhandensein von interstitiellen Abnormalitäten mit einer höheren Mortalität verbunden war.10 Derzeit ist jedoch unklar, wie Veränderungen identifiziert werden können, die zu einer IPF fortschreiten.
Fazit
Wie beim Lungenkarzinom handelt es sich auch bei der IPF um eine rauchassoziierte Erkrankung mit sehr schlechter Prognose. Das CT-Screening kann zwar nicht vor Lungenkrebs schützen, die Lungenkarzinomsterblichkeit wurde in der NLST-Studie jedoch um 20 % gesenkt. Um die Effizienz des CT-Screenings zu verbessern, könnte man im Rahmen des Lungenkrebs-Screenings bei Risikopatienten (Rauchanamnese) auch andere Krankheiten, wie eben die IPF, berücksichtigen.
Literatur:
1 Behr J et al., Dtsch Arztebl Int 2013; 110(51–52):875–81
2 American Thoracic Society ERS, Am J Respir Crit Care Med 2002; 165:277–304
3 American Cancer Society, ACS Cancer Facts and Figures 2009, 2009, Atlanta, GA
4 Vancheri C et al., Eur Respir J 2010; 35:496–504
5 Bjoraker JA et al., Am J Respir Crit Care Med 1998; 157:199–203
6 Kim DS et al., Proc Am Thorac Soc 2006; 3(4):285–92
7 Raghu G et al., Am J Respir Crit Care Med 2015; 192(2):e3–19
8 Aberle DR et al., NEJM 2011; 365(5):395–409
9 Jin GY et al., Radiology 2013; 268(2):563–71
10 Putman RK et al., JAMA 2016; 315(7):672–81
In Zusammenarbeit mit dem Fachmagazin Universum Innere Medizin

AutorIn: Priv.-Doz. Dr. Helmut Prosch
Universitätsklinik für Radiologie und Nuklearmedizin, Medizinische Universität Wien

Ursprünglich erschienen:
AEK 18|2017
AEK 18|2017