




















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Kardiale ATTR-Amyloidose
Frühzeitige Therapie verbessert Prognose
18. April 2025
Selten, undurchschaubar, schnell tödlich. Dieser Nimbus haftet der kardialen Amyloidose an – in der Mehrzahl der Fälle zu Unrecht, wie die Erfahrung zeigt. Durch klinische Aufmerksamkeit, Kenntnis der Pathomechanismen und die Fähigkeit, diese zum Wohl der Patient:innen in eine sinnvolle Therapie umzusetzen, kann zumindest die Transthyretin-(TTR-)assoziierte Amyloidose in der Mehrzahl der Fälle heute bereits oft gut behandelt werden.
Häufiger als gedacht
Vor allem in früheren Jahren wurde die kardiale ATTR-Amyloidose gemeinhin als „Zebra-Diagnose“ angesehen. Mittels neuer diagnostischer Möglichkeiten konnte jedoch gezeigt werden, dass deutlich mehr Patient:innen als bisher angenommen erkranken.1 Schätzungen gehen davon aus, dass über ein Zehntel der Patient:innen, die wegen einer Aortenstenose evaluiert werden, auch an kardialer ATTR-Amyloidose leiden könnten. In der Untergruppe von Patient:innen mit einer Low-Flow-low-Gradient-Aortenstenose dürfte dieser Anteil sogar bei knapp unter einem Drittel liegen.2 Über mehrere Studien mit Patient:innen mit „heart failure with preserved ejection fraction“ (HFpEF) hinweg lag der Anteil an Patient:innen mit kardialer ATTR-Amyloidose bei 11%.3
Verbesserte Prognose
Unter allen chronischen Erkrankungen des Myokardes ist die TTR-assoziierte Amyloid-Kardiomyopathie nach wie vor als eine der am stärksten malignen einzuschätzen – unbehandelt schwankt das mediane Überlebennach Erstdiagnose zwischen gut 24 bis hin zu knapp 70 Monaten je nach Krankheitsstadium.4 Durch spezifische Therapien sowie ein besseres Verständnis der Erkrankung ist die Wild-Type-TTR-Amyloid-Kardiomyopathie (d.h. ohne kausale genetische Variante im TTR-Gen) heute in aller Regel jedoch keine Erkrankung mehr, an der die Patient:innen deutlich verfrüht versterben. Dazu tragen neben dem durch spezifische Therapien protrahierten Verlauf auch ein sehr hohes Erstdiagnosealter und eine verbesserte Früherkennung bei.
Verbesserte Diagnostik
Dass die kardiale ATTR-Amyloidose heutzutage kaum mehr als Orphan Disease gewertet werden kann, liegt nicht zuletzt an deutlich verbesserten Werkzeugen in der Diagnostik. Während bis in die Mitte der 2010er-Jahre eine Biopsie des Herzmuskels notwendig war, kann die kardiale ATTR-Amyloidose heute durch eine Kombination aus bildgebenden (Ganzkörper-Knochenszintigrafie mit SPECT) und laborchemischen (Elektrophorese und Immunfixation aus Serum und Urin sowie die Quantifizierung der freien Leichtketten im Serum) Verfahren nachgewiesen werden (Abb.5).
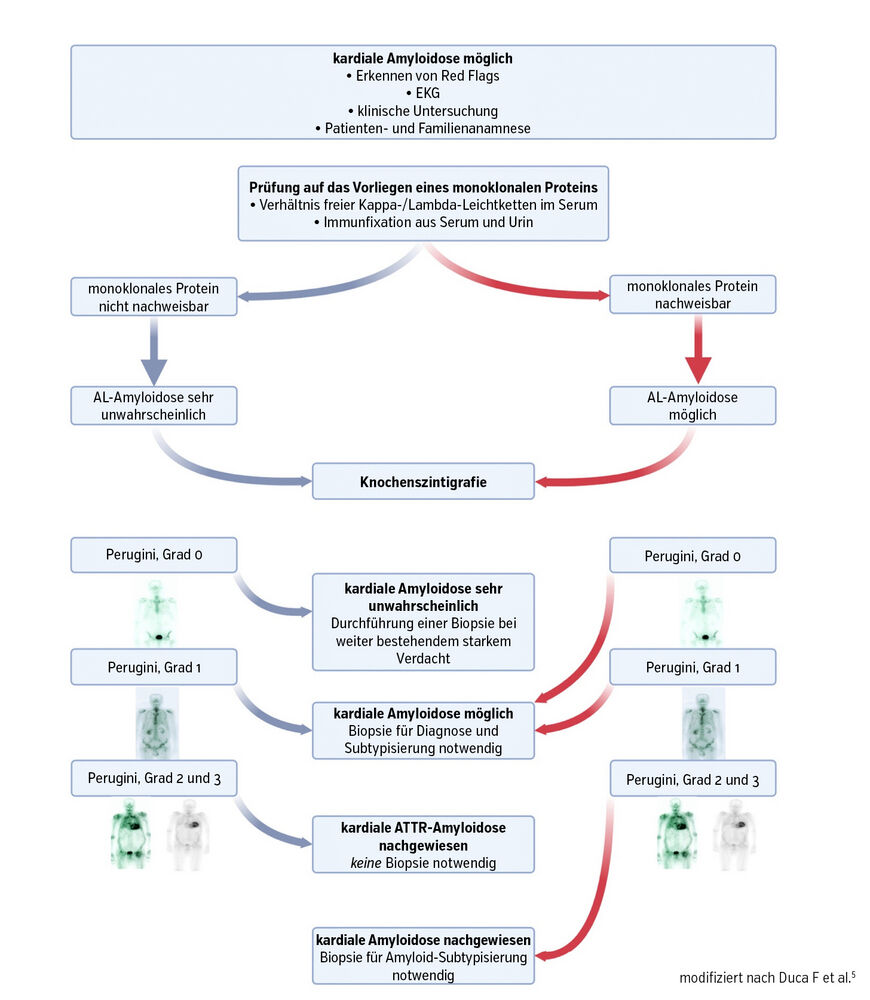
Abb.: Diagnostische Pfade bei kardialer Amyloidose
Wiewohl dieser Diagnoseweg gewisse Tücken aufweist und nicht auf alle Patient:innen gleichermaßen anwendbar ist, hat er dennoch geholfen, die Krankheit bei einer großen Anzahl an Patient:innen zu belegen und damit auch den Weg für die breite Etablierung wirksamer spezifischer Therapien zu ebnen.1 Essenziell ist dabei die Immunfixation aus Serum und Urin, um eine kardiale Leichtkettenamyloidose auszuschließen, die sich aus therapeutischer Sicht ganz wesentlich von der kardialen ATTR-Amyloidose unterscheidet.
Behandlung
Um Patient:innen mit kardialer ATTR-Amyloidose bestmöglich zu therapieren, hilft es, sich die pathophysiologischen und hämodynamischen Eigenschaften dieser Erkrankung vor Augen zu führen. Durch die Amyloid-Einlagerungen im Myokard ist dieses einerseits dicker und andererseits steifer – hämodynamisch handelt es sich also um eine restriktive Kardiomyopathie. Die Ventrikel sind dadurch kleiner, werfen pro Schlag selbst bei erhaltener Ejektionsfraktion weniger Blut aus und können sich bei körperlicher Belastung durch Erhöhung des Schlagvolumens nicht adäquat adaptieren, was als sogenannte inotrope Inkompetenz bezeichnet wird. Amyloid verändert nicht nur die mechanische Funktion des Myokardes, sondern beeinträchtigt in hohem Maße auch die elektrische Leitfähigkeit und damit das Reizleitungssystem des Herzens – die Folge davon können Rhythmusstörungen jeder Art sein.
Konsequenzen für Behandler:innen und Betroffene. Für die Therapie der kardialen ATTR-Amyloidose bedeutet dies, dass sowohl klinisch tätige Kardiolog:innen als auch Patient:innen selbst besonders auf zwei Basisfaktoren zu achten haben: (1) die engmaschige Kontrolle des Volumenhaushaltes beziehungsweise der eigenen Flüssigkeitsbilanz sowie (2) eine sorgfältige Überwachung des Herzrhythmus, um etwaige Störungen frühzeitig erkennen und gegebenenfalls gegensteuern zu können.
Spezifische Therapien. Als spezifische Therapien sind in Europa für die kardiale ATTR-Amyloidose die Wirkstoffe Tafamidis6 und Acoramidis7 zugelassen. Diese bewirken eine Stabilisierung der Transthyretin-Proteinstruktur im Blut und vermindern dadurch die zusätzliche Ablagerung von Amyloid.8 Es gilt jedoch zu beachten, dass die Therapie erst nach ungefähr 18 Monaten signifikante Überlebensvorteile bringt und bei Patient:innen mit einem noch nicht allzu fortgeschrittenen Krankheitsstadium tendenziell bessere Wirksamkeit zeigt.6 Effekte in Bezug auf die Lebensqualität und die Leistungsfähigkeit treten allerdings schon früher auf.6, 9 Wiewohl Transthyretin-Stabilisatoren aus der langfristigen Therapie der kardialen ATTR-Amyloidose nicht mehr wegzudenken sind, ist der höhere Gewinn an Lebensqualität und Leistungsfähigkeit für die Patient:innen im klinischen Alltag kurzfristig vor allem über die Einstellung des Volumenhaushaltes, das Rhythmusmanagement sowie das frühzeitige Erkennen und Abwenden kardialer Dekompensationen zu erreichen. In Zukunft werden neue und weiterentwickelte spezifische Therapeutika helfen, die Krankheitslast bei kardialer ATTR-Amyloidose weiter zu senken und in vielen Fällen ein nahezu normales Überleben zu ermöglichen und sie vielleicht sogar heilbar zu machen.7, 10
Conclusio
Für das optimale Management von Patient:innen mit kardialer ATTR-Amyloidose ist ein grundlegendes Verständnis der pathophysiologischen Vorgänge sowie ein rechtzeitiges Erkennen der Erkrankung essenziell. Insbesondere die wachsende Aufmerksamkeit für die Erkrankung wird in Zukunft helfen, noch mehr Patient:innen frühzeitig zu identifizieren und in einem frühen Krankheitsstadium einer spezifischen Therapie zuzuführen. Als inzwischen gut diagnostizier- und therapierbare Erkrankung ist die kardiale ATTR-Amyloidose aus dem Spektrum der praktisch relevantesten kardiologischen Erkrankungen jedenfalls kaum mehr wegzudenken.

Autor:
Dr. med. univ. Michael Poledniczek
Dr. med. univ. Michael Poledniczek
Klinische Abteilung für Kardiologie, Universitätsklinik für Innere Medizin II, Medizinische Universität Wien

Autor:
Priv.-Doz. Dr. Franz Duca, PhD
Priv.-Doz. Dr. Franz Duca, PhD
Klinische Abteilung für Kardiologie, Universitätsklinik für Innere Medizin II, Medizinische Universität Wien

Ursprünglich erschienen:
AEK 08|2025
AEK 08|2025
Literatur:
Ursprünglich erschienen in
UNIVERSUM INNERE MEDIZIN 5/24
UNIVERSUM INNERE MEDIZIN 5/24