





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Genetik der Epilepsien
12. Februar 2023
Epilepsiegenetik – ein Überblick
Anfallserkrankungen stellen sowohl von einem klinischen als auch von einem ätiologischen Standpunkt eine enorm heterogene Gruppe an Erkrankungen dar. Das mögliche ätiologische Spektrum ist breit und inkludiert erworbene bzw. strukturelle Ursachen wie ischämische Schlaganfälle, Blutungen, Tumoren, Enzephalitiden und Stoffwechselerkrankungen, aber auch genetische Syndrome wie z. B. die tuberöse Sklerose. Trotz detaillierter Ursachenforschung und mithilfe moderner diagnostischer Methoden lässt sich in 40–50 % der Fälle kein klarer Anfallsauslöser identifizieren, sodass diese Epilepsien als idiopathische oder nichtläsionelle Epilepsien bezeichnet werden. Aufgrund der familiären Häufung und der hohen Konkordanz bei monozygoten Zwillingen wird bereits seit langer Zeit auch für diese Gruppe von PatientInnen eine signifikante hereditäre Belastung angenommen.1 Im vergangenen Jahrzehnt konnte durch neue Forschungsmethoden die genetische Grundlage zahlreicher unterschiedlicher Anfallserkrankungen zunehmend entschlüsselt werden. Die verbesserte Kenntnis der genetischen Architektur hat im weiteren Verlauf dazu geführt, dass moderne Methoden nicht mehr ausschließlich als Forschungstool, sondern auch als Routinediagnostik Anwendung finden.
Der vorliegende Artikel soll einen kurzen Überblick über das genetische Spektrum der wichtigsten Epilepsien bereitstellen und beispielhaft aufzeigen, inwiefern die Kenntnis der zugrundeliegenden Genetik sowohl die Diagnostik als auch zunehmend das klinische Management revolutioniert hat.
Monogene Epilepsiesyndrome
Am offensichtlichsten erscheint der genetische Hintergrund der sogenannten monogenen Epilepsiesyndrome, bei denen eine genetische Veränderung (Mutation) in einem einzelnen Gen ausreicht, um eine Epilepsie hervorzurufen. Bis heute wurden mehrere hunderte Gene mit Epilepsien assoziiert, wovon die überwiegende Mehrheit in Familien mit solchen monogenen Epilepsien identifiziert wurde. So wurde im Jahr 1995 das erste Epilepsie-Gen, CHRNA4, das für einen Acetylcholin-Rezeptor kodiert, in einer Familie mit autosomal-dominant vererbter, nächtlicher Frontallappenepilepsie erstbeschrieben.2 Dadurch wurde die sogenannte „Channelopathy“-Ära eingeleitet, die durch die grundlegende Annahme charakterisiert war, dass der gemeinsame Pathomechanismus genetischer Epilepsien eine Dysfunktion von diversen Ionenkanälen sei. Den vermutlich bekanntesten Vertreter stellt das Natriumkanal-Gen SCN1A dar, das mit diversen Epilepsie-Phänotypen unterschiedlichen Schweregrades (darunter generalisierte Epilepsie mit Fieberkrämpfen, Dravet-Syndrom und fokale Epilepsien) in Verbindung gebracht wurde.3 Wie dieses Beispiel zeigt, können genetische Epilepsiesyndrome durch eine erhebliche Heterogenität gekennzeichnet sein. Das bedeutet einerseits, dass Veränderungen im gleichen Gen zu sehr unterschiedlichen klinischen Ausprägungen führen können (phänotypische Heterogenität) und andererseits, dass Veränderungen in verschiedenen Genen sehr ähnliche oder gar identische Phänotypen hervorrufen können (genetische Heterogenität).
Eine Revolution der epilepsiegenetischen Forschung wurde durch das Next-Generation Sequencing, also eine umfangreiche Hochdurchsatz-Sequenzierung kodierender Regionen des humanen Genoms, erreicht. Die Anwendung dieser Technologie führte zu einem exponentiellen Anstieg der Identifikation von Risikogenen für Anfallserkrankungen und veränderte auch unser Verständnis der Pathophysiologie. Initiale Bestrebungen betonten eine zentrale, aber komplexe Rolle zahlreicher unterschiedlicher Ionenkanal-Gene, welche auch heute noch eine funktionell bedeutende Gruppe an epilepsieassoziierten Genen darstellen.4 Dieses Konzept der Epilepsien als „Channelopathies“ wurde im weiteren Verlauf jedoch um zahlreiche Gene anderer funktioneller Gruppen erweitert, deren Proteinprodukte zum Beispiel beim neuronalen Zellwachstum eine essenzielle Bedeutung haben. Darunter fällt die Gruppe der drei Gene des GATOR1-Komplexes (DEPDC5, NPRL2, NPRL3), welcher im Normalzustand den mTOR-Signalpfad unterdrückt. Wird dieser supprimierende Mechanismus durch eine pathogene Mutation in einem dieser drei Gene aufgehoben, kann dies zu fokalen Epilepsien (nichtläsionell oder mit fokalen kortikalen Dysplasien) führen.5
Als spezielle Form des Next-Generation Sequencings führte speziell die Trio-Exom-Sequenzierung, bei der alle Exons (proteinkodierende Gensequenzen des gesamten Genoms) vom betroffenen Kind und beiden gesunden Eltern zur Identifizierung von Mutationen analysiert werden, die nicht im klassischen Sinne vererbt werden, sondern in der PatientInnengeneration neu entstanden sind (sogenannte De-novo-Mutationen). Durch diese Methode wurden vor allem bei schweren frühkindlichen Epilepsien mit konsekutiver Entwicklungsverzögerung oder -regression (epileptischen Enzephalopathien) zahlreiche neue Epilepsie-Gene entdeckt.6, 7
Monogene Epilepsien sind für sich stehend sehr seltene Erkrankungen und machen auch in ihrer Gesamtheit nur einen kleinen Anteil aller Epilepsien aus. Aufgrund des negativen (evolutionären) Selektionsdrucks sind auch die Mutationen, die solche monogenen Epilepsie-Syndrome verursachen, dementsprechend selten. Das Vorliegen der verantwortlichen Mutation führt in diesen Fällen allerdings mit einer sehr hohen Wahrscheinlichkeit (Penetranz) zum Ausbruch der Epilepsie. Grundsätzlich sollten dabei alle Mendel’schen Erbgänge (autosomal-rezessiv, autosomal-dominant inklusive De-novo-Mutationen, X-chromosomal) und auch die einzigartige Sonderform der X-chromosomalen Vererbung mit Aussparung des männlichen Geschlechts bei PCDH19-assoziierten Epilepsien, in Betracht gezogen werden. Das Wissen um die Erbgänge der jeweiligen Syndrome hat auch einen wesentlichen Einfluss auf eine adäquate genetische Beratung der betroffenen Familien.
Komplexe Vererbunghäufiger Epilepsien
Obwohl der Großteil der sogenannten idiopathischen (fokalen wie auch generalisierten) Epilepsien keine monogenen Erkrankungen darstellen, kann anhand von Zwillingsstudien und modernen genetischen Analysen eine starke genetische Komponente mit einer Gesamterblichkeit (Heritabilität) von etwa 70–80 % angenommen werden.8 Für diese Gruppe an häufigeren Epilepsie-Formen ist demgemäß eine multifaktorielle oder polygene Vererbung verantwortlich, bei der das Zusammenspiel von Veränderungen in zahlreichen Genen gemeinsam mit Umweltfaktoren für die Erkrankung prädisponiert. Diese Art der Vererbung lässt sich am besten durch das sogenannte Threshold-Modell (Abb. 1) veranschaulichen, nach dem die genetisch determinierte Anfallsbereitschaft (Suszeptibilität) einer Gauß’schen Normalverteilung folgt. Es ist dementsprechend davon auszugehen, dass eine Epilepsie erst dann manifest wird, wenn ein bestimmtes Ausmaß an genetischen und Umweltfaktoren in einem Individuum kumuliert, sodass schließlich eine kritische Schwelle an Risikofaktoren überschritten wird.9
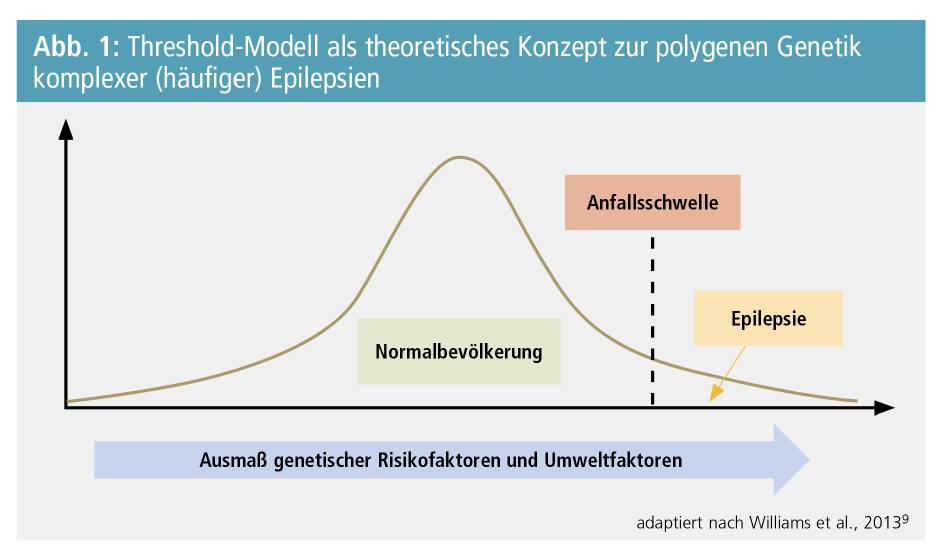
Es liegt folglich auch nahe, dass sich die Erfassung bzw. Quantifizierung eines solchen polygenen Hintergrundes deutlich komplexer als bei monogenen Erkrankungen darstellt. Eine anerkannte und seit vielen Jahren etablierte Methodik zur Identifizierung sogenannter Risikoloci in einem wissenschaftlichen Kontext stellen genomweite Assoziationsstudien (GWAS) dar, bei denen große Epilepsie-Kohorten (mit tausenden PatientInnen) mit einer noch größeren Anzahl an gesunden Kontrollen verglichen werden. Da diese genetischen Risikovarianten (wenn auch seltener) in gesunden Kontrollpopulationen auftreten, leisten sie einzeln (im Sinne einer Teilkausalität) einen verhältnismäßig geringen ätiologischen Beitrag zur Erkrankung und sind niemals alleine für das Auftreten einer Epilepsie verantwortlich. Erst durch die rasant wachsenden Kohortengrößen und dank internationaler Konsortien gelingt es, einen nennenswerten Anteil der Genetik komplexer Epilepsien auf diese Weise zu erklären. Eine rezent publizierte genomweite Metaanalyse zeigt klar, dass durch solche verhältnismäßig häufigen genetischen Veränderungen (auch Polymorphismen genannt) bereits über 30 % der Genetik erklärt werden kann, die den idiopathisch generalisierten Epilepsien zugrunde liegt.10 Auch bei fokalen Epilepsien und Fieberkrämpfen konnten über diesen Weg (allerdings in geringerem Ausmaß als bei generalisierten Epilepsien) signifikant mit den jeweiligen Erkrankungen assoziierte genetische Veränderungen identifiziert werden. Interessanterweise scheint es hier eine starke genetische Überlappung mit den seltenen monogenen Epilepsien zu geben, sodass sich bei Fieberkrämpfen und Temporallappenepilepsien unter anderem auch Niedrigrisiko-Varianten im SCN1A-Gen finden.11, 12
Zu diesem polygenen Konzept scheinen nach neuesten Erkenntnissen nicht nur häufige Polymorphismen, sondern auch sehr seltene Mutationen beizutragen, die in gesunden Kontrollen gar nicht vorkommen.13 Diese sind allerdings nicht über GWAS, sondern über Next-Generation Sequencing zu detektieren. Für die idiopathisch generalisierten Epilepsien dürfte hier speziell die Gruppe der GABAA-Rezeptor-Gene von besonderer Wichtigkeit sein.14
Klinisch-genetische Diagnostik bei Epilepsien
Genetisch-technologische Fortschritte der vergangenen Jahre haben die Detektionsrate pathogener Mutationen dramatisch verbessert, wodurch sich auch unser Verständnis der Biologie der Epilepsien nachhaltig verändert hat. Zunehmend werden dieselben Technologien nun auch in klinischen Laboratorien für diagnostische Zwecke eingesetzt. Für den Kliniker/die Klinikerin ist es daher von enormer Relevanz, die wesentlichen diagnostischen Methoden, die im klinischen Setting einsetzbar sind, mit all ihren Vor- und Nachteilen sowie den wichtigsten Indikationsgebieten zu kennen. Es gilt dabei festzuhalten, dass die althergebrachten Methoden mittlerweile weitgehend von modernen genomischen Techniken abgelöst worden sind.
Die früher häufig eingesetzte Einzelgenuntersuchung (mittels konventioneller Sanger-Sequenzierung) ist aufgrund der oft eingeschränkten Spezifität der meisten Epilepsiephänotypen nur mehr kaum indiziert. In Einzelfällen, bei denen die Klinik mit sehr hoher Wahrscheinlichkeit ein spezifisches Gen vermuten lässt (z. B. SCN1A beim Dravet-Syndrom, MECP2 beim Rett-Syndrom, SLC2A1 bei der GLUT1-Defizienz), ist diese Herangehensweise allerdings immer noch vertretbar. Auch die konventionelle Darstellung des Chromosomensatzes (Karyotypisierung) findet bei Epilepsien kaum Einsatz. Eine Ausnahme, die durch andere Methoden unzureichend erfasst werden kann, stellt das Ringchromosom 20 dar, an das vor allem bei therapierefraktären, lang andauernden Absenzen und nächtlichen tonischen Anfällen gedacht werden sollte.
Eine spezielle Art der potenziell mit Epilepsien assoziierten genetischen Variation sind sogenannte „Copy Number Variants“ (CNVs), also Deletionen und Duplikationen von subchromosomalen DNA-Abschnitten. Diese spielen vor allem bei syndromalen, schweren und frühkindlichen Epilepsien mit begleitender Entwicklungsverzögerung eine zentrale Rolle. Als Nachweismethode der Wahl steht hier der chromosomale Mikroarray zur Verfügung, durch den bei positivem Ergebnis eine klare Diagnose gestellt und das Wiederholungsrisiko gut abgeschätzt werden kann.
Die erhöhte Verfügbarkeit und fallende Kosten führen jedoch aktuell vermehrt dazu, dass das Next-Generation Sequencing immer früher im diagnostischen Prozedere zum Einsatz kommt. Hier stehen sehr unterschiedliche Applikationen von diversen Anbietern zur Verfügung. Das Hauptproblem für den klinischen Alltag ist, dass für die spezifische Auswahl keine klaren Richtlinien existieren. Aus diesem Grund richtet sich die Auswahl eher nach der Verfügbarkeit bzw. nach ökonomischen Aspekten (wie z. B. Kostenübernahme durch Krankenkassen). Einerseits können Genpanels angefordert werden, die sich voneinander vorwiegend im Ausmaß der sequenzierten/analysierten DNA unterscheiden. Kommerzielle Anbieter bieten dabei Genlisten an, die von wenigen Genen bis zu mehreren hunderten Genen („Comprehensive Epilepsy Panels“) reichen.
Eine gute Alternative hierzu stellt die Sequenzierung der gesamten kodierenden DNA-Sequenzen (Exom-Sequenzierung) dar. Ein klarer Vorteil dieser Anwendung ist, dass neue Gene detektiert und die Daten zu einem späteren Zeitpunkt auch im Hinblick auf zwischenzeitlich identifizierte Gene reanalysiert werden können. Der diagnostische Nutzen einer systematischen Reanalyse wurde kürzlich auch speziell für Epilepsien nachgewiesen.15 Allerdings muss bei der enorm großen Datenmenge, die durch die Exom-Sequenzierung generiert wird, vor der Überinterpretation von Varianten unklarer klinischer Signifikanz (VUS) gewarnt werden. PatientInnen müssen im Übrigen vor Durchführung einer Exom-Sequenzierung auch ausführlich darüber aufgeklärt werden, dass die Methode zur (nebenbefundlichen) Detektion pathogener Mutationen in Genen führen kann, die nichts mit der Epilepsie zu tun haben, aber dennoch von medizinischer Relevanz sind (z. B. Tumorprädispositionsgene wie BRCA1, APC). Für den Fall, dass eine De-novo-Mutation als Ursache vermutet wird, bietet sich speziell die bereits erwähnte Trio-Exom-Sequenzierung an, wobei gesunde Eltern und betroffenes Kind gemeinsam (als Trio) sequenziert werden und besonderes Augenmerk auf Varianten gelegt wird, die nur das Kind trägt.
Abbildung 2 schlägt einen Algorithmus zur genetisch-diagnostischen Abklärung bei Epilepsien vor, wobei explizit festzuhalten ist, dass aufgrund der immer niedriger werdenden Kosten und der verbesserten Interpretierbarkeit die Tendenz klar dahin geht, eine umfassende Diagnostik beispielsweise mittels Exom-Sequenzierung immer früher im Verlauf einzusetzen.
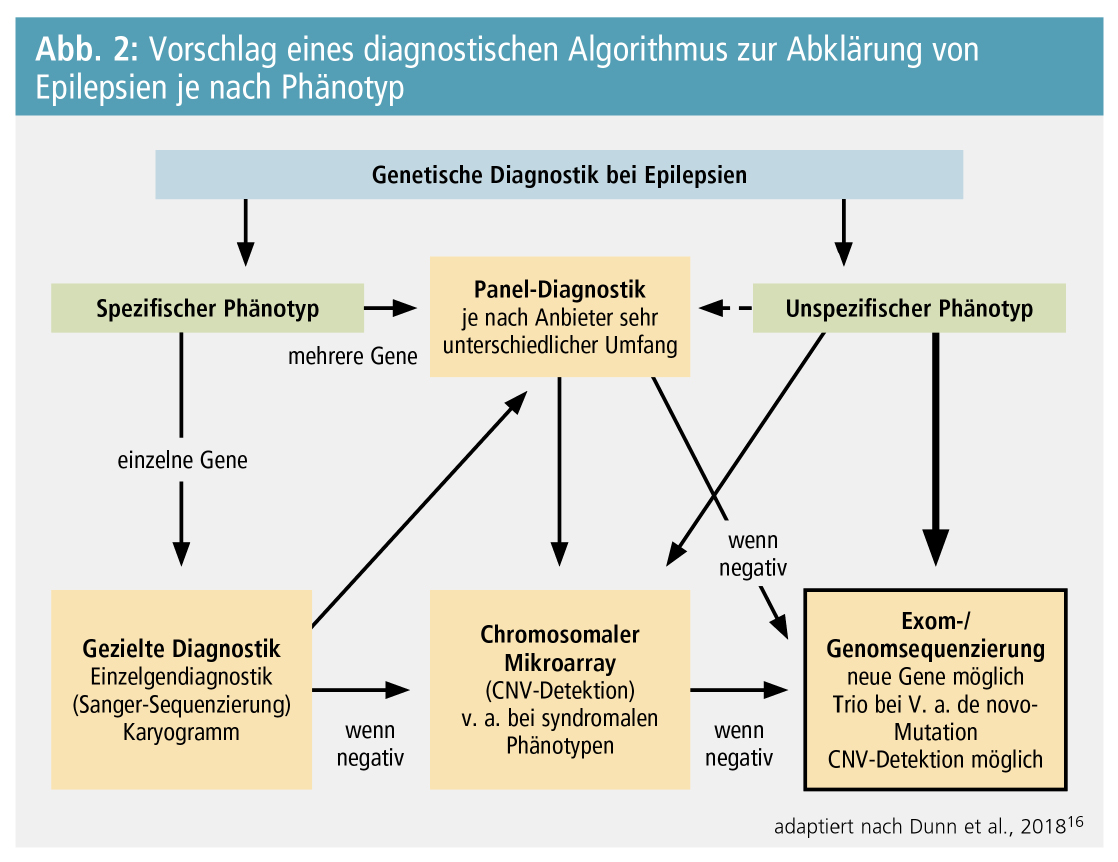
Zusammenfassend gilt es festzuhalten, dass eine genetische Diagnostik vorrangig für die schweren und syndromalen Epilepsieformen mit kombinierten Phänotypen (v. a. mit begleitender psychomotorischer Retardierung) in Erwägung gezogen werden soll. Bei häufigeren Epilepsien (v. a. idiopathische generalisierte und fokale Epilepsien) sollte eine Routinediagnostik nur dann erfolgen, wenn die Familienanamnese bzw. der Stammbaum für eine monogene Erkrankung spricht (Verwandte 1. Grades von ähnlichem Phänotyp betroffen).
Einfluss der Genetik auf die Therapie
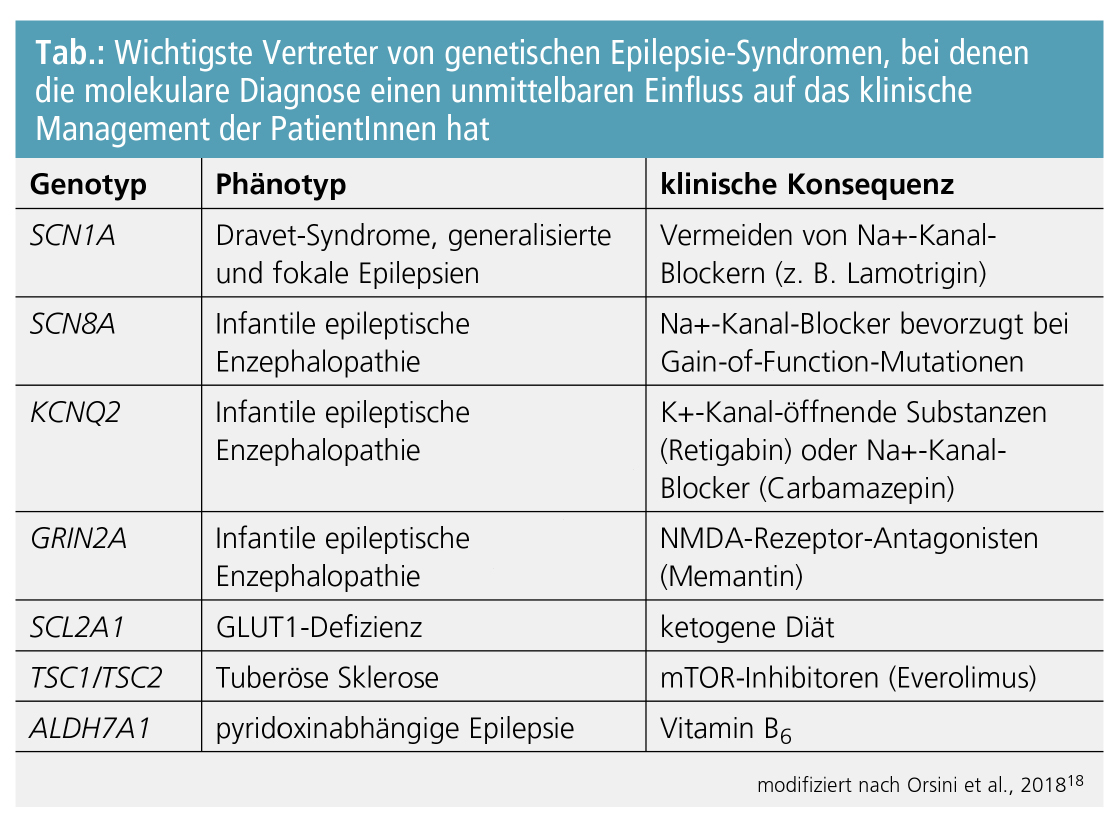
Rezente Entwicklungen in der Humangenetik haben vermehrt dazu geführt, dass zahlreiche Erkrankungen je nach nachgewiesener molekularer Ätiologie gezielt therapierbar wurden. Dieses Konzept der personalisierten Medizin hat auch Eingang in die Neurologie und im Speziellen auch in die Epileptologie gefunden, wobei dies aktuell v. a. pädiatrische Phänotypen betrifft. Ein nennenswertes Beispiel stellt hier die tuberöse Sklerose dar, die durch die Einführung von mTOR-Inhibitoren (Everolimus) zu einer kausal therapierbaren genetischen Erkrankung wurde.17 Ein weiterer wichtiger Vertreter ist die GLUT1-Defizienz, die durch Mutationen im Gen SLC2A1 hervorgerufen wird. An diese Erkrankung sollte vor allem bei epileptischen Enzephalopathien mit begleitenden paroxysmalen Dyskinesien gedacht werden. Eine frühe Diagnose ist besonders essenziell, da die Erkrankung auf eine ketogene Diät anspricht. Auch die Auswahl konventioneller Antikonvulsiva kann sich nach dem molekularen Defekt richten. Zum Beispiel sind bei Mutationen in Natriumkanalgenen, die zu einem Funktionsverlust des Proteins führen („loss-of-function mutations“), Na+-Kanal-blockierende Substanzen zu vermeiden, wohingegen bei Na+-Kanal-Mutationen, die einen funktionellen Zugewinn verursachen („gain-of-function mutations“), dieselben Medikamente besonders gut wirksam sind. Eine Auswahl an genetischen Diagnosen, die einen Einfluss auf das klinische Management der PatientInnen haben, findet sich in der Tabelle.
Entgegen der lange bestehenden Annahme, dass monogene Epilepsien nicht auf epilepsiechirurgische Eingriffe ansprechen, zeigen neue Fallserien von fokalen Epilepsien durch Mutationen in DEPDC5, NPRL2 und NPRL3 (GATOR1-Komplex), dass hier ein durchaus zufriedenstellendes postoperatives Outcome erreicht werden kann.5 Da bei GATOR1-Komplex-assoziierten Epilepsien eine Überaktivität des mTOR-Pfades angenommen wird, liegt hier (ähnlich wie bei der tuberösen Sklerose) auch eine Wirksamkeit von mTOR-Inhibitoren nahe. Zuverlässige Medikamentenstudien liegen für diese PatientInnen-Gruppe allerdings noch nicht vor.
Neben den diagnostischen und therapeutischen Neuerungen im Bereich der monogenen Epilepsien gibt es bei den häufigeren komplexen Epilepsien vor allem pharmakogenetische Bestreben, aus denen bereits genetisch-diagnostische Empfehlungen für den klinischen Alltag resultierten. So ist zum Beispiel die Testung auf bestimmte HLA-Genotypen (HLA-B*1502) bei Individuen asiatischer Herkunft bereits etabliert, um kutane Nebenwirkungen von Carbamazepin zu vermeiden.19
Zusammenfassung
Im vergangenen Jahrzehnt konnten vor allem dank der Next-Generation-Sequencing-Technologie massive Fortschritte in der epilepsiegenetischen Forschung erzielt werden. Dies führte nicht nur zu einem deutlich verbesserten Verständnis der Biologie von (polygenen wie auch monogenen) Anfallserkrankungen, sondern auch zu rasanten Fortschritten in der klinischen Diagnostik. Eine steigende Anzahl an genetischen Epilepsien ist dadurch im Sinne der personalisierten Medizin bereits einer gezielten Therapie zugänglich geworden. Aufgrund der stetigen Verbesserung genetischer Technologien und der wachsenden Datensätze ist anzunehmen, dass auch in den nächsten Jahren bemerkenswerte Erkenntnisse hinzukommen und zunehmend auch die klinische Praxis beeinflussen werden.

Autor:
Dr. Martin Krenn, PhD
Dr. Martin Krenn, PhD
Universitätsklinik für Neurologie, Medizinische Universität Wien

Ursprünglich erschienen:
neuro 02|2019
neuro 02|2019
Herausgeber: Österreichische Gesellschaft für Neurologie, Prim. Univ.-Prof. Mag. Dr. Eugen Trinka, FRCP, Präsident der ÖGN
Publikationsdatum: 2019-06-25
Zur Ausgabe »
Publikationsdatum: 2019-06-25
Zur Ausgabe »
Literatur
Bildnachweis
Vorschaubild: AdobeStock_560024803_sdecoret