





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Hoffnung für Patient:innen mit spinaler Muskelatrophie
12. Februar 2023
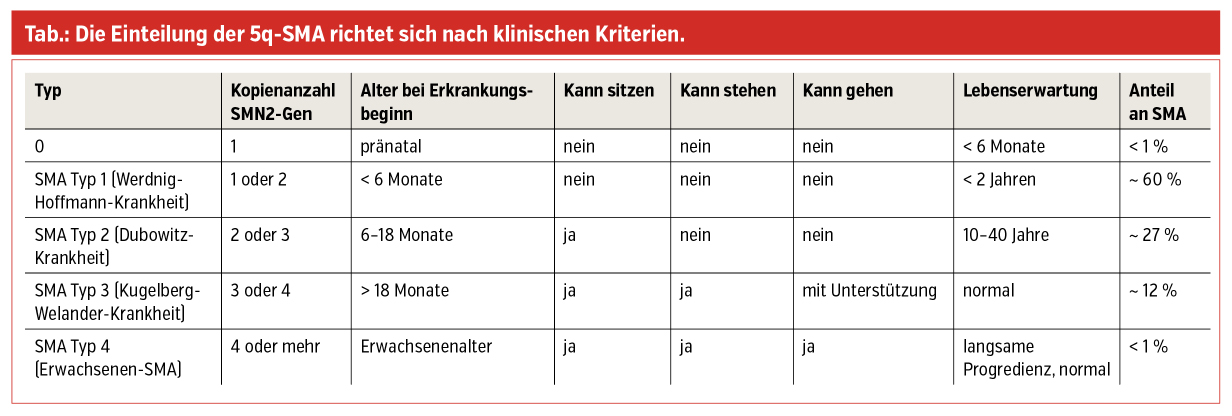
Die 5q-assoziierte spinale Muskelatrophie (SMA) stellt mit einer Inzidenz von 1 : 6.000–10.000 eine der häufigsten hereditären neuromuskulären Erkrankungen dar. Ursächlich ist eine homozygote Deletion im Survival-Motor-Neuron-(SMN-)1-Gen auf dem Chromosom 5q13, wodurch es zu einer Unterproduktion von funktionierendem SMN-Protein und in der Folge zu einer progredienten Degeneration der Alpha-Motoneuronen im Rückenmark kommt. Das SMN2-Gen am zentromerischen Teil des Chromosom 5 unterscheidet sich vom SMN1-Gen nur durch wenige Basenpaare. Durch das fehlende Exon 7 wird das SMN2-Gen jedoch zum Großteil nicht transkribiert, was zu einem nicht funktionierenden bzw. rasch abgebauten SMN-Protein führt. Aus lediglich 20 % des SMN2-Transkripts entsteht ein voll funktionsfähiges Protein. Die Krankheitsausprägung wird daher überwiegend durch die individuell unterschiedliche Anzahl der SMN2-Gen-Kopien beeinflusst. Allen Typen gemeinsam ist eine fortschreitende, symmetrische, proximal betonte Muskelschwäche, bei der die Beine früher und stärker betroffen sind als die Arme, sowie fehlende oder deutlich abgeschwächte Muskeleigenreflexe. Eine kognitive Einschränkung besteht nicht. Die SMA Typ 1 stellt mit 60 % die häufigste und (nach Typ 0) schwerste Form der Erkrankung dar und war bis vor Einführung der neuen Therapien vor wenigen Jahren die häufigste Todesursache im Säuglings- und Klein-kindalter.
Kausale Therapie
Die wachsende Kenntnis über die Pathophysiologie der SMA ermöglichte die Entwicklung dreier kausal orientierter Medikamente, deren gemeinsames Ziel die Herstellung eines voll funktionsfähigen SMN-Proteins ist. Nusinersen (Spinraza®), ein Antisense-Oligonukleotid, modifiziert das Splicen der inkompletten Information des SMN2-Gens und wurde 2017 als erstes Medikament von der Europäischen Arzneimittelbehörde (EMA) für alle Altersgruppen, Phänotypen und Erkrankungsstadien zugelassen. Nach einer Aufsättigungsphase muss es alle 4 Monate intrathekal verabreicht werden.
Im Mai 2020 folgte die Zulassung der Genersatztherapie mit Onasemnogene-Abeparvovec (Zolgensma®) für SMA-Betroffene mit max. 3 SMN2-Genkopien und einem Gewicht von bis zu 21 Kilogramm. Dabei wird intakte SMN1-DNA über einen adenoassoziierten viralen Vektor (AAV9) einmalig intravenös verabreicht. Seit 2021 ist auch das Small Molecule Ridiplam (Evrysdi®) in Europa zugelassen, das wie Nusinersen das Splicen der inkompletten Information des SMN2-Gens modifiziert und für alle Altersgruppen und Schweregrade der Erkrankung zugelassen ist. Es muss täglich oral eingenommen werden. In den Zulassungsstudien und Veröffentlichungen mit Real-Life-Daten konnte für diese drei Medikamente eine hervorragende Wirksamkeit bei gutem Nutzen-Risiko-Verhältnis bewiesen und gezeigt werden, dass ein früher, am besten präsymptomatischer Beginn der Therapie eine grundlegende Prognoseänderung und bestenfalls Heilung bedeutet.
Daher wurde das österreichische Neugeborenenscreeningprogramm 2021 um SMA erweitert. Bereits 14 Patient:innen konnten so präsymptomatisch detektiert und einer Therapie zugeführt werden. Die seit 2007 publizierten Standards of Care (SoC) stellen weiterhin als Versorgungsrichtlinien die Basis für eine optimale Patientenbetreuung hinsichtlich Langzeitmanagement und interdisziplinäre symptomatische Therapie dar.

Autorin:
Dr.in Anna Hüpper
Dr.in Anna Hüpper

Autor:
Prim. Univ.-Prof. Dr. Günther Bernert
Prim. Univ.-Prof. Dr. Günther Bernert
Präsident Österreichische Muskelforschung, Neuropädiater

Ursprünglich erschienen:
AEK 03|2023
AEK 03|2023
Praxistipps
-
Die Diagnose SMA sollte bei jedem Säugling mit rumpfbetonter (proximaler) Hypotonie vermutet werden. Zusätzliche Hinweise in jedem Alter sind motorische Schwierig-keiten, Verlust von motorischen Fähigkeiten, proximale Muskelschwäche, Hypo- oder Areflexie und Zungenfaszikulationen.
-
Bei V. a. Muskelschwäche ist der Ausschluss einer SMA notwendig, da im positiven Fall jede Therapieverzögerung zum weiteren Fortschreiten des Kraftverlusts führt („Time is motoneuron“).
-
Die Einhaltung der Standards of Care (SoC) und die Anbindung an ein neuromuskuläres Zentrum oder eine Spezialambulanz sind essenziell, um Komplikationen zu vermeiden und das bestmögliche individuelle Outcome zu erreichen.
Was Patient:innen wissen möchten
Ist eine Heilung mit den neuen Therapien möglich?
Die Krankheitsdauer vor Behandlungsbeginn und die Anzahl der SMN2-Genkopien sind der wichtigste prognostische Faktor und bestimmen den individuellen Krankheitsverlauf. Für alle SMA-Phänotypen ist mit den neuen medikamentösen Therapieoptionen eine klare Verbesserung des Outcomes zu erwarten.
Gibt es Unterschiede in der Wirksamkeit zwischen den einzelnen genbasierten Therapien?
Bisher konnten keine Unterschiede belegt werden.
Bildnachweis
Vorschaubild: AdobeStock_433023570_Dr_Microbe