





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Immunschwäche bei Abwehrstörungen: Eine frühe Erkennung ist wichtig!
18. Juni 2021
Primäre Abwehrstörungen werden als primäre Immundefekte (PID) oder seit kurzem auch als „inborn errors of immunity“ (IEI) bezeichnet. In diesen Überbegriffen werden seltene angeborene Erkrankungen des Immunsystems zusammengefasst, deren Systematik jedes 2. Jahr in einem Konsensus-Report der internationalen Gesellschaften für Immunologie herausgegeben wird. Darin wird sowohl auf die genetischen Grundlagen als auch auf die phänotypischen Krankheitsmerkmale eingegangen. Ein alle 2 Jahre stattfindendes Update ist notwendig, da der medizinische Fortschritt in diesem Feld besonders schnell ist; so wurden z. B. zwischen dem letzten Report und dem noch gültigen aus dem Jahr 2019 über 60 neue Krankheitsbilder entdeckt. PID werden in 10 Gruppen unterteilt; die prominenteste Gruppe stellen die Antikörperdefekte dar, es gibt jedoch auch Krankheiten, die man nicht a priori zu den Immundefekten zählen würde, z. B. das familiäre Mittelmeerfieber (Gruppe: autoinflammatorische Erkrankungen) oder Syndrome mit multiplen Autoimmunerkrankungen (Gruppe: Immundysregulation).
Dieses umfassende Klassifikationssystem untermauert auch die große Bandbreite der Symptome von primären Immun-defekten.
Die Prävalenz von PID ist unterschiedlich und sehr von der geografischen Lage abhängig. So kommt z. B. der IgA-Mangel als häufigste angeborene Antikörperimmunschwäche mit einer Prävalenz von 1 : 163 in Spanien, jedoch mit nur 1 : 14.840 in Japan bei asymptomatischen Populationen vor. Die Prävalenz von symptomatischen Immundefekten in europäischen Registern wird mit 4/100.000 angenommen.
Die meisten PID haben eine genetische Ursache und zeigen sich vorwiegend im frühen Kindesalter. Es gibt auch Erkrankungen, die erst im Erwachsenenalter manifest werden. Dies sind meist Störungen der Antikörperbildung, wie z. B. die CVID („common variable immunodeficiency“), die sich im Median erst um das 24. Lebensjahr manifestiert.
Zur besseren klinischen Erkennung von PID wurde eine AWMF-Leitlinie der Arbeitsgemeinschaft Pädiatrische Immunologie und der Deutschen Gesellschaft für Immunologie herausgegeben, die zuletzt 2017 aktualisiert wurde.
Die hier gezeigten Akronyme sind dieser Leitlinie entlehnt.
Klinische Diagnose eines Immundefekts bei Infektanfälligkeit
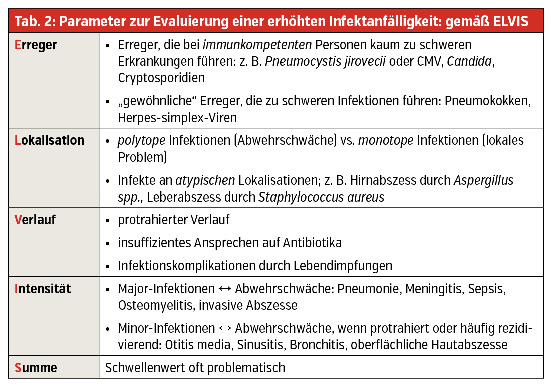
Bei den meisten primären Immundefekten ist das Leitsymptom eine erhöhte Infektanfälligkeit. Da dies oft vom subjektiven Empfinden der Patienten sowie von der jeweiligen Lebenssituation abhängt, wurde mehrmals der Versuch einer objektiven Definition übernommen. Im deutschen Sprachraum hat sich hier das Akronym ELVIS (Erreger, Lokalisation, Verlauf, Intensität, Summe) zur Einschätzung der Infektneigung durchgesetzt.
Bezüglich der Summe der Infektionen muss festgehalten werden, dass zwar wegen der subjektiven Komponente des Krankheitsgefühls ein Schwellenwert problematisch ist, dass aber die europäische Gesellschaft für Immundefekte einen Richtwert von 4 oder mehr antibiotikapflichtige Infekte (Otitis, Bronchitis, Sinusitis, Pneumonie) angibt.
Klinische Diagnose eines Immundefekts bei Störung der Immunregulation
Eine gestörte Immunregulation kann manchmal auch alleiniges Symptom von PID sein. Auch hier wurde ein Akronym gefunden: GARFIELD (Granulome, Autoimmunität, rezidivierendes Fieber, ekzematöse Hauterkrankungen, Lymphoproliferation, chronische Darmentzündung).
Labordiagnostik eines Immundefekts
Bei klinischem Verdacht auf einen Immundefekt sollte man eine Stufendiagnostik (Basisdiagnostik und in weiterer Folge auch spezifische Antikörper) durchführen (Tab. 1).
Auf einen Immundefekt lassen eine Lymphozytopenie, Neutropenie (absolut, nicht relativ) sowie Monozytopenien, Thrombozytopenien oder eine Eosinophilie (im Rahmen einer Immundysregulation) schließen.

Eine Antikörpermangelerkrankung zeigt sich in einer variablen Senkung der IgA-, IgM-und IgG-Spiegel inklusive der IgG-Subklassen, in anderen Fällen ist jedoch auch ein erhöhter Titer von IgE oder IgM möglich.
Bei Persistenz des Verdachts trotz normaler Werte sollten in spezialisierten Zentren zusätzliche Untersuchungen stattfinden, wie z. B. die Antikörpertiter nach einer Impfung, eine Quantifizierung und Charakterisierung von B- und T-Zellen sowie funktionelle Assays von T-Zellen und/oder Phagozyten.
In weiterer Folge kann eine genetische Diagnostik angeschlossen werden. Aufgrund von variabler Expression und Penetranz kann es jedoch bei verschiedenen Patienten trotz gleicher Mutation zu unterschiedlichen klinischen Phänotypen kommen. Daher sollte ein genetischer Befund nur in Zusammenhang mit klinischer Symptomatik und restlichen Befunden von diesbezüglich erfahrenen Ärzten interpretiert werden.
Therapie
Die Betreuung von PID-Patienten sollte auf jeden Fall in dafür spezialisierten Zentren erfolgen, deren es in Österreich mehrere gibt.
Die Therapie von PID basiert auf 4 Pfeilern:
- der antibiotischen Prophylaxe,
- der Substitution von Immunglobulinen,
- der Therapie der Autoimmunphänomene mit Immunsuppressiva und
- dem Versuch, das mutierte Gen auszuschalten – entweder durch moderne Gentechnik oder durch eine Stammzelltransplantation.
Während eine Antibiotikaprophylaxe (z. B. mit Sulfamethoxazol/Trimethoprim oder Azithromycin) niederschwellig gegeben werden kann, sollte die Indikation für die Gabe von Immunglobulinen und Immunsuppressiva von Spezialisten gestellt werden. Stammzelltransplantationen oder Gentherapien werden nur in spezialisierten Zentren für sonst letale (meist kombinierte zelluläre) PID durchgeführt.

Wissenswertes für die Praxis
- Bei klinischem Verdacht auf einen PID sollte bei erhöhter Infektanfälligkeit gemäß ELVIS (Tab. 2) und bei Verdacht auf Störung der Immunregulation gemäß GARFIELD vorgegangen werden (Tab 3).
- Die Diagnostik sollte in einem Stufenprinzip durchgeführt werden (Tab 1).
- Die Therapie basiert auf 4 Säulen.
PID-Zentren in Österreich
Wien Kinder
- Medizinische Universität Wien, Universitätsklinik für Kinder- und Jugendheilkunde, Ambulanz für Störungen der Immunabwehr
- Klinik Donaustadt, Abteilung für Kinder- und Jugendheilkunde, Rheumatalogische Immunologische und Hämatologische Spezialambulanz
Wien Erwachsene
- AKH, Rheuma-Ambulanz mit integrierter PID-Ambulanz für Erwachsene
- Hanusch-Krankenhaus, Rheuma-Ambulanz mit integrierter PID-Ambulanz für Erwachsene
- Immunologische Tagesklinik für Erwachsene
Innsbruck
- Medizinische Universität Innsbruck, Universitätsklinik für Innere Medizin II, Ambulanz für Infektiologie, Immunologie, Tropenmedizin
Graz
- Landeskrankenhaus-Universitätsklinikum Graz, Klinische Abteilung für Rheumatologie und Immunologie, Immunologische Ambulanz

AutorIn: Prof. Priv.-Doz. Dr. Ruth D. E. Fritsch-Stork
Sigmund Freud Privatuniversität Wien, PID-Ambulanz der 1. Medizinische Abteilung, Hanusch-Krankenhaus, Wien
© Gerhard Krejci

Ursprünglich erschienen:
AEK 12|2021
AEK 12|2021