





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
RNAi-Technologie
Therapie der hereditären Transthyretin-Amyloidose
20. Oktober 2023
Standen noch vor wenigen Jahren keine spezifischen Wirkstoffe zur Verfügung, so wandelte sich die ATTRv durch revolutionäre molekulare Technologien von einer unheilbaren zu einer behandelbaren Erkrankung.
Pathophysiologie
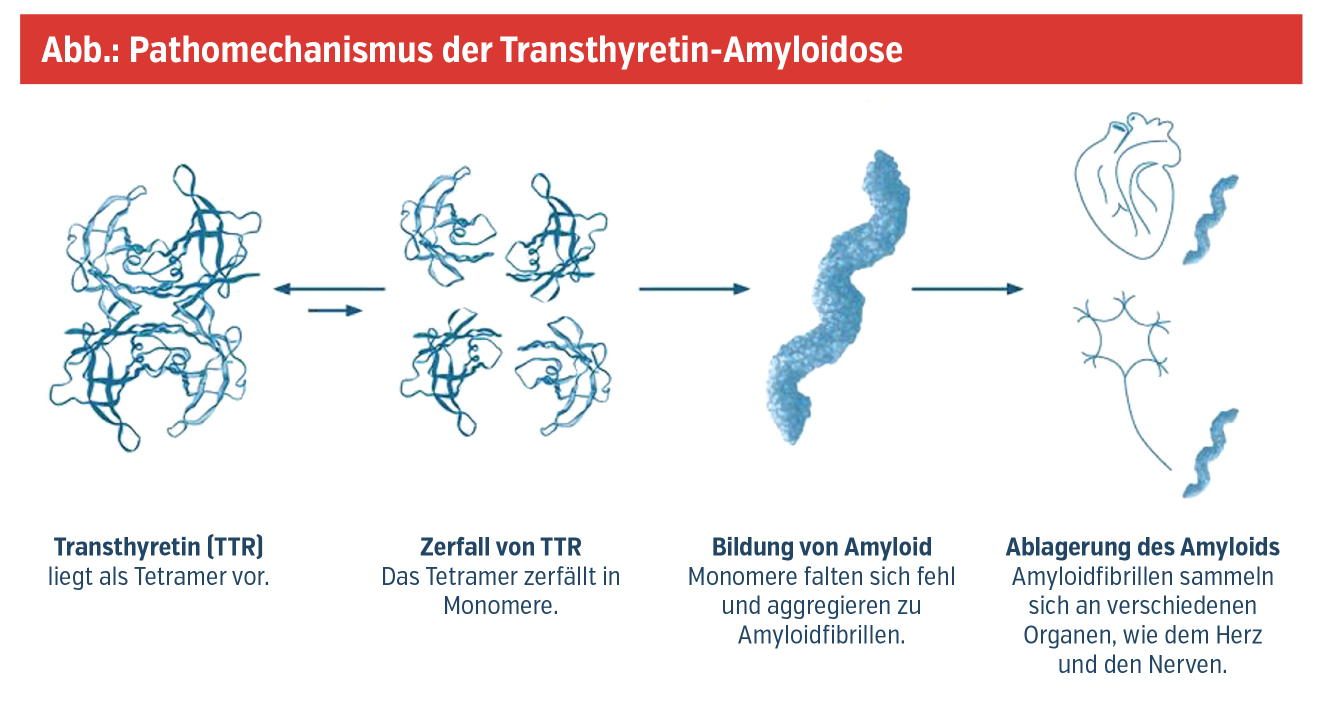
Als Amyloide bezeichnet man fehlgefaltete Proteine, die sich durch enzymatische Umwandlungsprozesse als unlösliche Komplexe in Form von Amyloidfibrillen in diversen Organen ablagern und so zu deren Dysfunktion im Sinne der Erkrankung Amyloidose führen. Die Internationale Amyloidose-Gesellschaft listet derzeit mehr als 30 Proteine, die beim Menschen eine Amyloidose verursachen können. Zu der häufigsten und somit klinisch relevantesten Form der Amyloidosen zählt die Transthyretin-Amyloidose (ATTR), deren Pathogenese von Transthyretin (TTR), einem hepatisch synthetisierten Transportprotein für Vitamin A und Schilddrüsenhormone, dominiert wird. Im Rahmen der Erkrankung lässt sich eine nichterblich bedingte, altersassoziierte Wildtypform (ATTRwt) von einer autosomal-dominant vererbbaren, hereditären (varianten) Form (ATTRv) abgrenzen. Während die Pathogenese der ATTRwt noch nicht vollständig geklärt ist, kommt es bei der ATTRv bedingt durch Punktmutationen im TTR-Gen zur Destabilisierung des TTR-Tetramers und Dissoziation in seine Monomere, die schließlich Amyloidfibrillen formen (Abb.).
Klinische Präsentation
Die klinische Präsentation der Patient:innen hängt naturgemäß davon ab, welche Organe von den Amyloidablagerungen betroffen sind, wobei sich ATTR-Ablagerungen überwiegend in Herz und/oder peripherem Nervensystem manifestieren. Beim kardiologischen Phänotyp kommt es aufgrund der myokardialen Amyloidablagerung und der daraus resultierenden erhöhten Myokardsteifigkeit zur Ausprägung einer Herzinsuffizienz mit vorwiegend diastolischer Dysfunktion. Diese ist durch Belastungsdyspnoe, Beinödeme, Hepatomegalie, Aszites und einen erhöhten Jugularvenendruck gekennzeichnet.
Der neurologische Phänotyp der ATTRv ist durch die unterschiedliche Verteilung der Ablagerungen bei unterschiedlichen Mutationen relativ heterogen, jedoch primär von sensorischer Polyneuropathie (PNP) und autonomer Dysregulation geprägt. Die Erkrankung beginnt meist mit sensiblen Neuropathien in den Zehen und breitet sich proximal aus, wobei in weiterer Folge motorische Funktionen rasch betroffen sein können. Charakteristisch zeigt sich eine ausgeprägte Dysautonomie, mit Symptomen wie Dyshidrose, sexuelle Dysfunktion oder gastrointestinale Beschwerden. Zu den weiteren häufigen Manifestationen zählt ein oftmals bilaterales Karpaltunnelsyndrom.
Diagnose
Der wichtigste Schritt in der Diagnosestellung einer ATTR liegt darin, die Erkrankung differenzialdiagnostisch in Betracht zu ziehen, wobei für eine korrekte und rasche Diagnostik eine gute interdisziplinäre Zusammenarbeit essenziell erscheint.
Die diagnostische Abklärung einer kardialen ATTR wird neben der klinischen Präsentation und laborchemischen Parametern primär von bildgebenden Untersuchungen dominiert. Dabei nimmt neben der Echokardiografie und der kardialen Magnetresonanztomografie vor allem die Ganzkörperknochenszintigrafie eine Schlüsselrolle in der nichtinvasiven Diagnostik ein. Serum- und Harnanalyse bilden eine weitere diagnostische Säule. In dieser kann Paraprotein in den entsprechenden Immunfixationen sowie eine pathologische Kappa-Lambda-Ratio im Serum hinweisend auf eine leichtkettenassoziierte Amyloidose (AL) sein, die differenzialdiagnostisch jedenfalls in Betracht gezogen werden sollte. Aufgrund des breiten Spektrums an diagnostischen Möglichkeiten ist heutzutage eine korrekte Diagnose in den meisten Fällen auch ohne Endomyokardbiopsie möglich. Bei unklaren Befundkonstellationen oder speziellen Fragestellungen gilt die Herzmuskelbiopsie jedoch nach wie vor als Goldstandard in der Amyloidosediagnostik.
Sofern primär neurologische Symptome vorherrschend sind, steht zur weiteren diagnostischen Abklärung hinsichtlich vorliegender PNP neben der Messung der Nervenleitgeschwindigkeit auch die Elektromyografie zur Verfügung. Weiters kann zur Diagnosestellung eine Biopsie aus subkutanem Fettgewebe, Speicheldrüsen, Rektum oder betroffenen Nerven erfolgen. Eine negative Biopsie schließt jedoch das Vorliegen einer ATTRv nicht aus. Die genetische Analyse des TTR-Gens sollte in jedem Fall auch bei gesicherter ATTR-Diagnose zur weiteren Abklärung erfolgen, um zu unterscheiden, ob es sich dabei um eine ATTRv oder ATTRwt handelt. Daraus lassen sich wiederum unterschiedliche therapeutische Maßnahmen ableiten.
Spezifische Therapieoptionen
Historisch gesehen stellte bei der Diagnose einer ATTRv eine Transplantation der betroffenen Organe bzw. der Leber als TTR-Produktionsort die einzige therapeutische Option dar. Zunehmende Awareness und reges Forschungsinteresse führten in den letzten Jahren jedoch zu weitreichenden Entwicklungen hinsichtlich therapeutischer Optionen. Standen noch vor wenigen Jahren keine spezifischen Wirkstoffe zur Verfügung, so sind neben TTR-Stabilisatoren (Tafamidis) seit 2018 die 2 Präparate Patisiran und Inotersen zur Behandlung der ATTRv-assoziierten PNP zugelassen (Kasten). Seit September 2022 wurde die Palette um Vutrisiran erweitert. Eplontersen, eine Weiterentwicklung von Inotersen, wird derzeit noch in einer Phase-III-Studie untersucht. Die Wirksamkeit und Sicherheit von Vutrisiran bzw. Eplontersen werden außerdem sowohl bei Patient:innen mit ATTRv als auch ATTRwt mit kardialer Beteiligung geprüft. Die Verschreibung erfolgt durch Fachärzt:innen für Neurologie, die Behandlung sollte von einem/einer in der Therapie von Patient:innen mit Amyloidose erfahrenen Mediziner:in eingeleitet und überwacht werden.

Autor:
Dr. René Rettl
Dr. René Rettl

Autorin:
Assoz. Prof.in Priv.-Doz.in Dr.in Diana Bonderman
Assoz. Prof.in Priv.-Doz.in Dr.in Diana Bonderman
5. Medizinische Abteilung mit Kardiologie und internistische Notaufnahme, Klinik Favoriten, Wien

Ursprünglich erschienen:
AEK 20|2023
AEK 20|2023
Neue molekulartechnologische Therapieoptionen
- Patisiran (Onpattro®, i. v.): Mittels Ribonukleinsäure-Interferenz (RNAi) können Gene stillgelegt (Gen-Silencing) werden. Durch Spaltung der mRNA wird die zu übertragende Information zerstört und in weiterer Folge die Translation in ein Protein gehemmt. Patisiran supprimiert die TTR-Produktion in den Hepatozyten und senkt so die Serum-TTR-Konzentration um bis zu 80 %, wodurch Bildung und Ablagerung von Amyloidfibrillen in den Organen deutlich reduziert werden. In der Zulassungsstudie wurde bei den behandelten Patient:innen eine Verbesserung der PNP festgestellt, während jene in der Placebogruppe eine Progedienz ihrer Erkrankung erlitten.
- Inotersen (Tegsedi®, s. c.): Durch spezifische Bindung mittels Antisense-Oligonukleotiden an eine komplementäre mRNA („Antisense“-Technologie) wird durch Hemmung der Translation und RNA-Interferenz die Produktion von TTR in den Hepatozyten supprimiert. Auch Inotersen konnte die Serum-TTR-Konzentration um 75–79 % senken. Es zeigte sich eine geringere Progredienz der PNP in der Behandlungsgruppe im Vergleich zu Placebo.
- Vutrisiran (Amvuttra®, s. c.): Neue Formulierung von Patisiran, seit September 2022 in der EU zugelassen. Auch mit Vutrisiran konnte eine Verbesserung der PNP festgestellt werden. Zeigte im Vergleich zu Patisiran weniger Nebenwirkungen und bietet den Vorteil der subkutanen Verabreichung (alle 3 Monate statt alle 3 Wochen i. v.).
Literatur bei den Verfasser:innen
Ursprünglich erschienen in
UNIVERSUM INNERE MEDIZIN 04|2021
Themenheft Seltene Erkrankungen
UNIVERSUM INNERE MEDIZIN 04|2021
Themenheft Seltene Erkrankungen