





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Der Begriff „Kardiomyopathie“ beschreibt allgemein eine Erkrankung des Herzmuskels, die durch strukturelle oder funktionelle Abnormitäten auffällt. Bei allen Formen kommt es zu einer Veränderung der Struktur des Herzmuskelgewebes. Aufgrund der vielfältigen Präsentation – sowohl aufgrund der unterschiedlichen Ätiologie als auch der Ausprägung, die sich im Laufe der Erkrankung auch ändern kann – ist eine klinisch sinnvolle Einteilung eine Herausforderung. Regelmäßig wurden von nationalen und internationalen Fachgesellschaften neue Definitionen und Klassifikationsschemata bereitgestellt. Aus historischen Gründen werden die ischämische, valvuläre, kongenitale sowie hypertensive Kardiomyopathie, bei denen die Ursache bekannt zu sein scheint, unter dem engeren Begriff der Kardiomyopathien nicht abgehandelt. Im Jahr 2023 wurde nun von der Europäischen Gesellschaft für Kardiologie (ESC) die erste internationale alle Formen umfassende Leitlinie zum Management der Kardiomyopathien veröffentlicht. Kardiomyopathien können klinisch unauffällig sein und nur zufällig entdeckt werden oder sich primär durch eine Arrhythmie oder Herzinsuffizienz manifestieren. Die Gründe für die Erstvorstellung bewegen sich dementsprechend auf einem weiten Spektrum. Die Patient:innen können aufgrund eines Zufallsbefundes in der Bildgebung elektrokardiografische Auffälligkeiten, kardiale Symptome, manifeste Herzinsuffizienz, Herzrhythmusstörungen bis zum plötzlichen Herztod, aber auch wegen auffälliger Ergebnisse beim Familienscreening bei Angehörigen oder aufgrund einer Abklärung bei einer Multiorganerkrankung zugewiesen werden. Im Verlauf schreitet die Erkrankung immer weiter voran, mit teils schwerwiegenden Verlaufsformen. Bei unter 55-jährigen Menschen sind Kardiomyopathien die Hauptursache für eine Herztransplantation.
Die Diagnose Kardiomyopathie sollte unbedingt die Suche nach ihrer Ursache zur Folge haben. Das Wissen über die Ursache hat Auswirkungen auf die Patientenaufklärung, die Prognoseabschätzung, die Bewertung hinsichtlich des Risikos für plötzlichen Herztod (SCD), das generelle Management und in einigen speziellen Fällen auch auf die Therapie. Bei familiären Formen spielt auch die Indikationsstellung zum genetischen Testen sowie die Früherkennung oder Therapie der betroffenen Familienmitglieder eine wichtige Rolle.
Am Beginn des diagnostischen Pfades steht die Beurteilung des Phänotyps (Tab.).
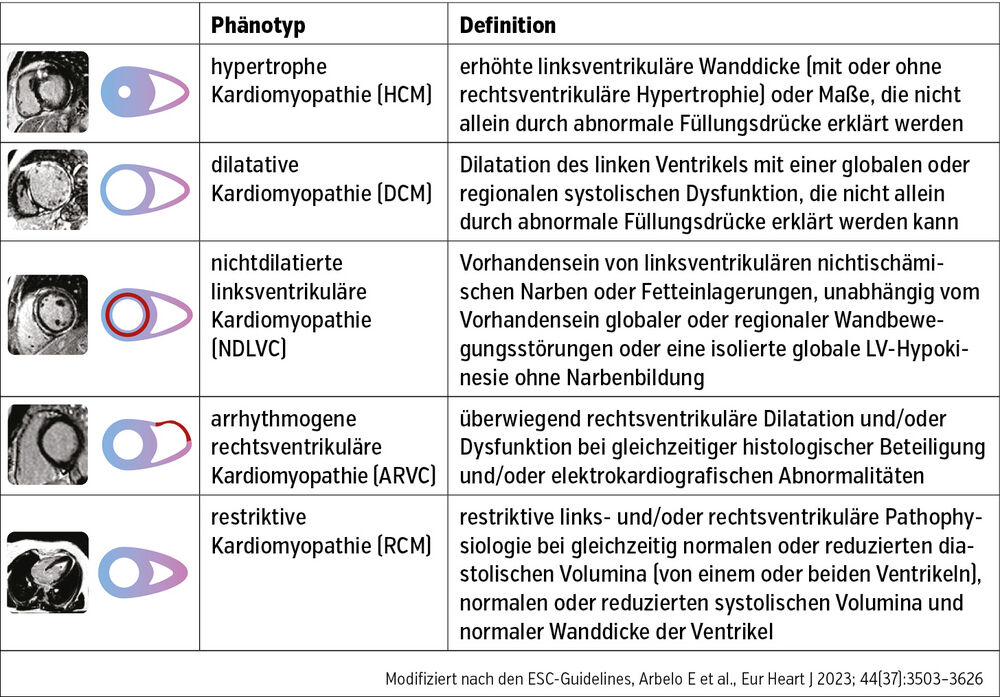
Tab.: Klassifikation bei Kardiomyopathien: der Phänotyp. Erster Schritt bei der Suche nach der Ätiologie
Der Phänotyp und eventuelle klinische Red Flags helfen bei der Priorisierung der Diagnostik, da einige der Ursachen mehr oder weniger wahrscheinlich sind. Man kann auch zwischen primären (z. B. genetische Formen, Myokarditis) und sekundären Kardiomyopathien (z. B. als Teil einer Systemerkrankung) unterscheiden. Einige Formen wie die hypertrophe Kardiomyopathie zeigen eine starke Assoziation ihrer Ausprägung und ihres Verlaufs mit einzelnen genetischen Mutationen. Jedoch dürfte ein bestimmter genetischer Hintergrund den meisten Formen im Sinne einer erhöhten Suszeptibilität oder Second-Hit-Theorie zugrunde liegen.
Therapiemöglichkeiten
Generell gelten für die Kardiomyopathien bei entsprechender Klinik die Behandlungsrichtlinien der Herzinsuffizienz und der Arrhythmien. Steht eine Herzinsuffizienz mit eingeschränkter Pumpfunktion im Vordergrund, sollten alle hier evidenzbasierten Therapien verschrieben werden (4-Säulen-Konzept). Ein wichtiger Punkt bei Kardiomyopathien stellt die Bewertung des SCD-Risikos dar. Bei erhöhtem Risiko wie etwa bei Vorliegen von Hochrisiko-Mutationen kann eine primär prophylaktische ICD-(Implantierbarer-Kardioverter-Defibrillator-)Implantation außerhalb des klassischen Indikationsbereiches, also auch bei einer LVEF >35 %, erfolgen. Bei sekundären Kardiomyopathien, also Mitbeteiligung des Herzens im Rahmen von anderen Erkrankungen, steht neben der kardialen Therapie die Behandlung der Grunderkrankung im Fokus. Intensive körperliche Belastungen sollten in einigen selektierten Patientengruppen vermieden werden, aber in den meisten Fällen ist eine moderate körperliche Betätigung in Absprache mit den behandelnden Ärzt:innen möglich und wird sogar empfohlen.
Zur Zeit wird intensiv an Therapien für spezifische Kardiomyopathien geforscht und seltene Formen in internationalen Registern gesammelt, um das Wissen zu erweitern. Große Fortschritte konnten bereits in der pharmakologischen Therapie bei der kardialen ATTR-Amyloidose, einer infiltrativen Kardiomyopathie (TTR-Stabilisatoren wie Tafamidis, Synthesereduktion durch RNA-Silencing wie Patisiran und Vutrisiran oder Antisense-Oligonukleotid wie Inotersen und zukünftig eventuell „Depleter“-Antikörper) sowie der hypertrophen obstruktiven Kardiomyopathie (kardiale Myosin-Inhibitoren wie Mavacamten und Aficamten) erzielt werden.
Praxismemo
- Bei einer strukturellen oder funktionellen Abnormität des Herzens sollte stets die Ursache abgeklärt werden.
- Das kardiale MRT ermöglicht eine funktionelle Untersuchung und Gewebecharakterisierung und gibt somit wichtige Hinweise zur Ätiologie.
- Für die kardiale ATTR-Amyloidose und hypertrophe obstruktive Kardiomyopathie sind spezifische Therapien verfügbar.

Autorin:
Ap. Prof.in Priv.-Doz.in Dipl.-Ing.in Dr.in Noemi Pavo, PhD
Ap. Prof.in Priv.-Doz.in Dipl.-Ing.in Dr.in Noemi Pavo, PhD
Klinische Abteilung für Kardiologie, Universitätsklinik für Innere Medizin II, Medizinische Universität Wien

Ursprünglich erschienen:
AEK 12|2024
AEK 12|2024
SCHWIERIGE PROGNOSE
Aufgrund der bemerkenswerten genetischen Variabilität der Kardiomyopathien und unterschiedlicher äußerer Einflüsse ist eine Vorhersage, ob, wann, in welcher Form und mit welchem Verlauf eine Erkrankung auftritt, nicht genau möglich. Bei Vorliegen von Hochrisiko-Mutationen kann eine prophylaktische ICD-Implantation erfolgen.
DILATATIVE KARDIOMYOPATHIE (DCM)
Bei einer DCM sind die Herzkammern und die Herzvorhöfe erweitert, die Patient:innen leiden an der typischen Form einer Herzschwäche. Bei etwa der Hälfte der Fälle kann keine Ursache gefunden werden. Für das Management gelten die Leitlinien für die Herzinsuffizienz und die implantierbaren Geräte entsprechend den ESC-Empfehlungen.
HYPERTROPHE KARDIOMYOPATHIE (HCM)
Bei der HCM ist der Herzmuskel verdickt, wobei meistens die linke Herzkammer betroffen ist. Es werden eine obstruktive und eine nichtobstruktive Form unterschieden. Etwa 2 Drittel lassen sich durch eine monogene Ursache erklären. Bei jungen, oftmals sportlichen Menschen ist sie die häufigste Ursache für den plötzlichen Herztod.
WAS PATIENT:INNEN WISSEN WOLLEN
Wie lange muss ich die medikamentöse Therapie einnehmen?
Fast ausschließlich alle Formen der Kardiomyopathien stellen eine chronische Erkrankung dar. Deswegen müssen die unterstützenden medikamentösen Therapien lebenslang eingenommen werden, um das Voranschreiten der Erkrankung zu verlangsamen und dadurch das Leben zu verlängern. Nur in den allerseltensten Fällen tritt eine kardiale Funktionsstörung vorübergehend auf und „heilt aus“ (z. B. einige Fälle von Herzmuskelentzündung, Stress-Kardiomyopathie).
Muss ich eine genetische Untersuchung machen, bzw. was kann ich mir davon erwarten?
Die Leitlinien empfehlen ein genetisches Testen, wenn davon ein Nutzen für den/die Patient:in selbst oder für Angehörige zu erwarten ist. Dies kann die Sicherung/den Ausschluss einer genetischen Diagnose sein, aber in seltenen Fällen auch einen direkten Einfluss auf die Behandlung bzw. die Behandlung von Angehörigen haben. Die Indikationsstellung erfolgt immer nach einem gemeinsamen Konsensus im ärztlichen Gespräch.
Literatur bei der Verfasserin
Bildnachweis
Vorschaubild: © Kanisorn – stock.adobe.com