






















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Hypertroph-obstruktive Kardiomyopathie
Aufbruch in eine neue Ära
5. Juli 2024
Die hypertrophe Kardiomyopathie (HCM) ist im Erwachsenenalter definiert als eine meist im Herzultraschall gemessene Wanddicke im linken Ventrikel (LV) ≥ 15 mm an einer beliebigen Stelle, großteils im interventrikulären Septum, ohne abnorm veränderte Druckverhältnisse wie beispielsweise bei unbehandeltem Bluthochdruck oder Aortenklappenstenosen. Die HCM wird in zwei Gruppen eingeteilt: eine nichtobstruktive Form und eine obstruktive Form (HOCM), bei der an mindestens einer Stelle im LV ein Druckgradient ≥ 30 mmHg gemessen wird, meist im linksventrikulären Ausflusstrakt (LVOT).
Epidemiologie und Differenzialdiagnosen
Die Prävalenz liegt bei ca. 1 : 500, womit die HCM die häufigste genetische Kardiomyopathie darstellt, die einem autosomal dominanten Erbgang folgt. Die Mutationen betreffen meist Proteine des kardialen Sarkomers, jedoch kann der Phänotyp der HCM auch ohne genetische Mutation vorliegen bzw. durch andere Entitäten wie beispielsweise eine kardiale Amyloidose oder einen Morbus Fabry hervorgerufen werden. Diese alternativen Krankheitsbilder gehören durch ein umfangreiches diagnostisches Work-up initial ausgeschlossen, da sie konsekutiv eine gänzlich unterschiedliche Therapie benötigen.
Therapieoptionen
Viele der betroffenen Patient: innen sind asymptomatisch, weshalb die Diagnose der HCM meist als Zufallsbefund erfolgt. Führende Beschwerden symptomatischer Patient:innen sind typische Belastungsdyspnoe sowie Angina-Pectoris-Beschwerden.
Obstruktionsmanagement
Besteht eine signifikante Obstruktion des LVOT bei symptomatischen HOCM-Patient:innen, empfehlen die aktuellen Richtlinien der Europäischen Gesellschaft für Kardiologie (ESC) eine Erstlinientherapie mit Beta-Blockern oder Nicht-Dihydropyridin-Kalziumkanalblockern. Sollte trotz maximal tolerierter Dosis eine entsprechende Symptomatik bestehen, ist in weiterer Folge eine Therapie mit dem ersten spezifischen Therapeutikum in Erwägung zu ziehen. Es handelt sich bei der Substanz Mavacamten um einen selektiven, reversiblen Myosininhibitor, der aktiv in den Kontraktionsablauf eingreift, indem die vorherrschende Hyperkontraktilität abgeschwächt und somit der LVOT-Gradient reduziert wird.
Die Zulassungsstudie EXPLORER-HCM konnte auf beeindruckende Weise zeigen, dass Patient:innen mit Mavacamten sowohl von der subjektiven Symptomatik als auch anhand objektivierbarer Parameter (maximale Sauerstoffaufnahme, maximaler LVOT-Gradient) signifikant besser abschnitten als die Patient:innen in der Placebogruppe. Erfreulicherweise fand sich auch in den Langzeitdaten der Erweiterungsstudien keine relevante Einschränkung der linksventrikulären Pumpfunktion.
Weiters konnte in der Studie VALOR-HCM gezeigt werden, dass nach entsprechender Mavacamten-Therapie signifikant weniger Patient:innen für eine invasive Therapie wie die katheterinterventionelle Alkoholablation oder die chirurgische Myektomie geeignet waren im Vergleich zu Placebo. Diese Ergebnisse spiegeln sich auch in den aktuellen Leitlinien wider, wonach eine Mavacamten-Therapie im Therapiealgorithmus noch vor den invasiven Verfahren in Betracht gezogen werden soll (Abb.). Eine wichtige Voraussetzung für die Einleitung dieser Therapie ist u. a. eine linksventrikuläre Auswurffraktion > 55 %.
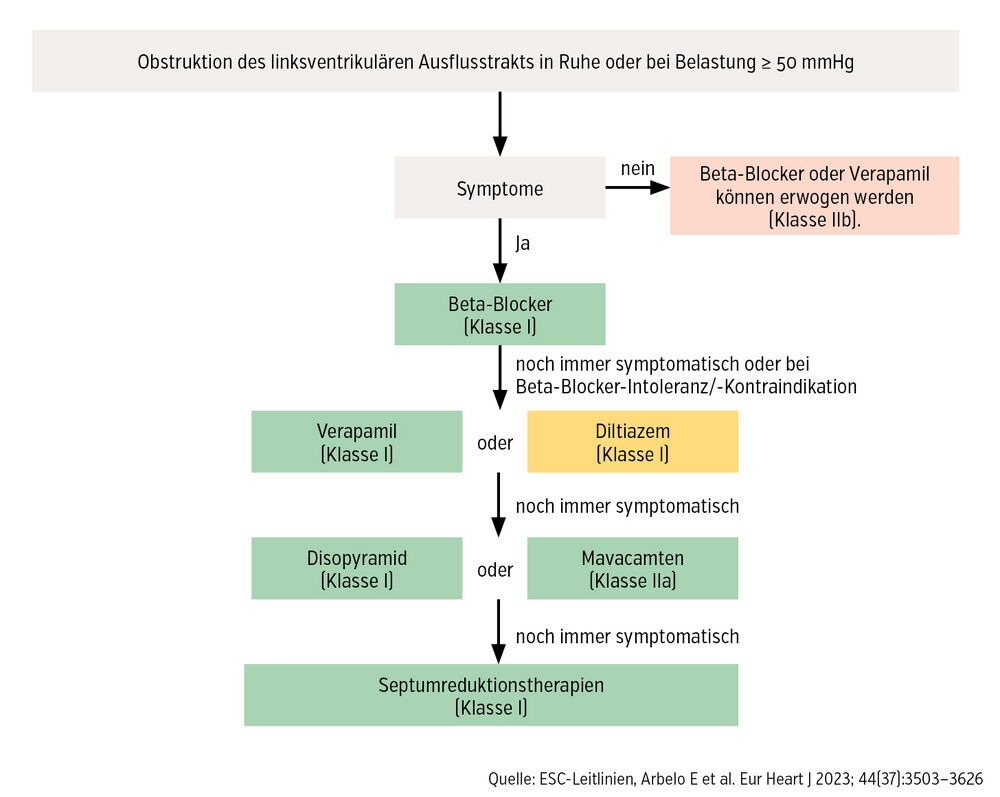
Abb.: Therapiealgorithmus zur Behandlung der hypertroph-obstruktiven Kardiomyopathie
Risikostratifizierung
Ein weiterer wesentlicher Bestandteil im Management der HCM ist die regelmäßige Risikostratifizierung hinsichtlich des plötzlichen Herztodes. In den meisten Studien zeigte sich das jährliche Risiko bei 1–2% aller HCM-Patient:innen. Während die führenden Arrhythmien zweifellos spontanes Kammerflimmern bzw. ventrikuläre Tachykardien darstellen, können auch höhergradige AV-Blockierungen bzw. spontane Asystolien auftreten. Sowohl die amerikanische als auch die europäische kardiologische Gesellschaft haben Scores entwickelt, die anhand anamnestischer, klinischer und bildgebender Parameter ein entsprechendes 5-Jahres-Risiko für den plötzlichen Herztod aufzeigen. Auf Basis dieser Wahrscheinlichkeit wird individuell die Dringlichkeit bzw. Notwendigkeit einer primärprophylaktischen Defibrillatorimplantation entschieden. Ob eine aktive Therapie mit Mavacamten das Arrhythmierisiko langfristig reduzieren kann, ist Gegenstand laufender Studien und bleibt eine spannende Frage für die Zukunft.
Genetische Testung
Obwohl ein genetischer Genotyp nicht zwangsläufig mit der Ausprägung eines klinischen Phänotyps vergesellschaftet sein muss, hat das genetische Testen in den aktuellen europäischen Richtlinien eine enorme Aufwertung erfahren. Vorausgesetzt, dass kompetente genetische Beratung mit entsprechender Expertise sichergestellt werden kann, soll eine genetische Testung sowohl zur Diagnosesicherung als auch zum familiären Screening bei einer bereits gesicherten Mutation eines Familienmitglieds durchgeführt werden.

Autor:
Priv.-Doz. DDr. Daniel Dalos
Priv.-Doz. DDr. Daniel Dalos
Universitätsklinik für Innere Medizin II, Klinische Abteilung für Kardiologie,
Medizinische Universität Wien

Ursprünglich erschienen:
AEK 13-14|2024
AEK 13-14|2024
WISSENSWERTES FÜR DIE PRAXIS
- Bis zu 2 Drittel aller Patient:innen weisen entweder in Ruhe oder unter Belastung/Valsalva-Manöver eine signifikante LVOT-Obstruktion auf.
- Gerade bei unklarer Symptomatik oder eingeschränkter Bildqualität sollte ein Belastungstest bzw. eine Dobutamin-Echokardiografie zur Detektion eines LVOT-Gradienten erfolgen.
- Sowohl in der Differenzialdiagnostik als auch in der Risikostratifizierung hat die kardiale Magnetresonanztomografie einen essenziellen Stellenwert.
- Nach Etablierung einer Mavacamten-Therapie (Einleitungsphase) muss innerhalb der ersten 3 Monate alle 4 Wochen eine Kontroll-Echokardiografie zur Bestimmung der Pumpfunktion erfolgen.
- Mavacamten soll nicht bei schweren Nieren- oder Leberfunktionsstörungen begonnen werden, und es muss besonders auf das Interaktionspotenzial (z. B. Protonenpumpenhemmer) geachtet werden.
WAS PATIENT:INNEN WISSEN WOLLEN
Brauche ich einen Defibrillator?
Die Diagnose HCM bedeutet nicht automatisch, dass Sie einen Defibrillator benötigen. Ob Ihr Risiko für gefährliche Rhythmusstörungen erhöht ist, wird anhand diverser Untersuchung von Ihrem/Ihrer Ärzt:in in regelmäßigen Abständen festgelegt.
Wie oft muss ich zur Kontrolle kommen?
Die Kontrollintervalle richten sich nach Ihrer Symptomatik. Wenn Ihre Symptome mit entsprechender Therapie (medikamentös/invasiv) kontrolliert sind, werden 1-mal jährliche Kontrollen empfohlen.
Literatur beim Verfasser
Bildnachweis
Vorschaubild: © Kanisorn – stock.adobe.com