





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Multiple Sklerose und verwandte Erkrankungen
Therapiefortschritte in der Neuroimmunologie
10. Mai 2024
Die multiple Sklerose (MS) ist eine chronisch entzündliche Erkrankung des zentralen Nervensystems (ZNS) und der häufigste Vertreter im Spektrum der entzündlichen demyelinisierenden ZNS-Erkrankungen. Die zwei wichtigsten Differenzialdiagnosen sind die Neuromyelitis-optica-Spektrum-Erkrankungen (NMOSD) und die MOG-Antikörper-assoziierten Erkrankungen (MOGAD). Da die MS vorwiegend junge Erwachsene betrifft, ein hohes Risiko für Behinderung birgt und effektive Therapieoptionen zur Verfügung stehen, ist eine frühzeitige Diagnose essenziell.
Das klinische Bild der MS weist hohe interindividuelle Unterschiede hinsichtlich Schweregrades, Verlaufs und Symptomatik auf. Bei ca. 85% beginnt die MS schubförmig (RMS) mit zwischenzeitlicher Remission. Schübe können je nach Lokalisation der ZNS-Läsion zu unterschiedlicher sowohl monofokaler als auch multifokaler Symptomatik wie Visus-Störung, Paresen oder Sensibilitätsstörungen führen. Frauen sind 3-mal häufiger betroffen. Die Häufigkeit von Schüben variiert stark und beträgt im Schnitt unbehandelt 0,5/Jahr. Bei der primär progredienten Verlaufsform (PPMS) steht eine langsam progrediente Symptomatik im Vordergrund, zumeist in Form einer zunehmenden spastischen Para-/Tetraparese. Bei NMOSD gibt es nur schubförmige Verläufe. Typische Zeichen sind rezidivierende Optikusneuritiden (ON) und Myelitiden. Weitere Syndrome sind das Area-postrema-Syndrom (Singultus, Nausea, Vomitus) oder eine symptomatische Narkolepsie. Schübe verlaufen deutlich schwerer als bei der MS. Ein fast vollständiger Visusverlust oder eine bilaterale Beteiligung des Sehnerves stellen klassische Befunde dar, wie sie bei MS nahezu nie vorkommen. Ebenso weist eine langstreckige Myelitis (longitudinale extensive transverse Myelitis, LETM) mit ausgeprägten bilateralen Paresen und einer schweren Blasenentleerungsstörung auf eine NMOSD hin (MS: partielle kurzstreckige Myelitis). Ohne aggressive Behandlung mit Glukokortikoiden und früher Plasmapherese ist das Risiko für einen schweren Residualzustand bereits nach dem ersten Schub hoch.
Durch die Entdeckung der Myelin-Oligodendrozyten-Glykoprotein-(MOG-)Antikörper ließ sich in den letzten Jahren eine neue Krankheitsentität abgrenzen (MOGAD). Das klinische Bild ist variabler als bei NMOSD, und Kinder sind häufiger betroffen als Erwachsene. Bei Kindern ist eine akute disseminierte Enzephalomyelitis (ADEM) typisch, während bei Erwachsene häufiger eine ON oder LETM vorliegt. Eine Besonderheit ist das Überwiegen monophasischer Verläufe. Das Risiko eines multiphasischen Verlaufs liegt zwischen 35–50%.
Diagnose
Bei typischer Anamnese und Klinik soll eine weitere Abklärung eingeleitet werden (Abb.).
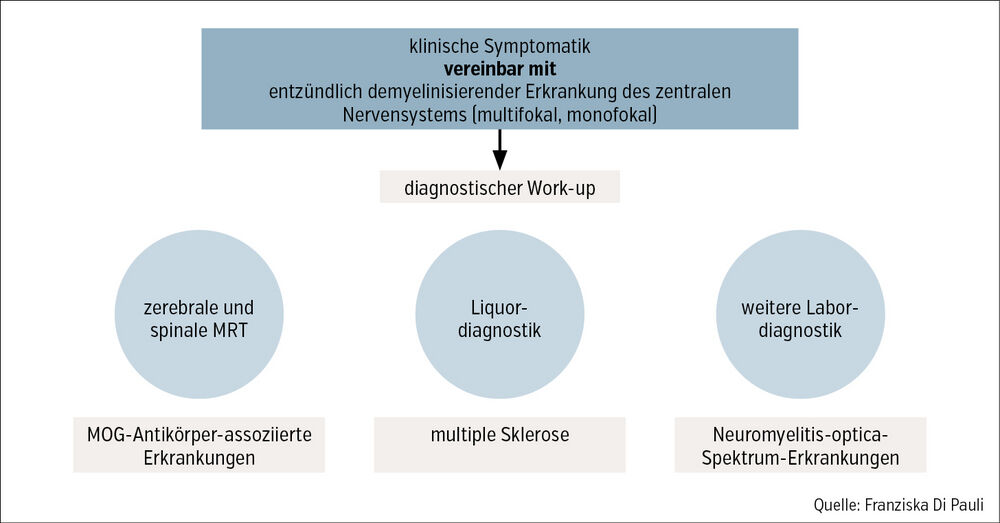
Abb.: Algorithmus zur Diagnose bei klinischem Verdacht auf entzündlich demyelinisierende Erkrankung des zentralen Nervensystems
Für Diagnose (und Differenzialdiagnose) spielen die Magnetresonanztomografie (MRT) und die Liquordiagnostik die größte Rolle. Die Diagnose der MS erfolgt anhand der revidierten McDonald-Kriterien 2017, wobei im Rahmen eines ersten klinischen Ereignisses der Nachweis zeitlicher und räumlicher Dissemination gefordert ist. Dies ist anhand der MRT für die räumliche (mehrere MS-typische Läsionen an zumindest 2 von 4 Lokalisationen, d.h. [juxta]kortikal, periventrikulär, infratentoriell, spinal) und zeitliche Dissemination (paralleles Vorkommen von Kontrastmittel-[KM-] bzw. Nicht-KM-aufnehmenden Läsionen oder neue Läsionen im zeitlichen Verlauf) oder mittels Liquordiagnostik für die zeitliche Dissemination (positive oligoklonale Banden im Liquor) möglich.Da sich bei NMOSD und MOGAD Behandlungsstrategien und Krankheitsverlauf unterscheiden, ist eine frühe Abgrenzung von einer MS erforderlich. Bei klinisch atypischen Zeichen für MS bzw. typischer Klinik oder Bildgebung für NMOSD oder MOGAD sollten daher Aquaporin-4-(AQP-4-)Antikörper bzw. MOG-Antikörper im Serum bestimmt werden.
Therapie
Die Therapie in der Neuroimmunologie hat in den letzten Jahrzehnten enorme Fortschritte gemacht. Die Behandlung beruht auf den Säulen:
- akuter Schub,
- krankheitsmodifizierende Therapie (DMT) und
- symptomatische Maßnahmen.
Bei MS stehen bezüglich DMT verschiedene Präparate (> 20) zur Verfügung. Diese unterscheiden sich hinsichtlich immunmodulatorischen und immunsuppressiven Potenzials, Effektivität, Nebenwirkungen und Risiken sowie Applikationsform. Die Auswahl hängt von Krankheitsaktivität, Krankheitsverlauf und individuellen Merkmalen wie z.B. Familienplanung ab.
Bei der NMOSD erfordert bereits die erste klinische Manifestation eine unmittelbare DMT. Seit kurzem stehen 3 Präparate (Eculizumab, Satralizumab, Inebilizumab), die in verblindeten placebokontrollierten Phase-III-Studien für wirksam befunden wurden, für die AQP-4-Antikörper-positive NMOSD zur Verfügung.
Ein anderes Therapiekonzept kommt bei MOGAD zur Anwendung. Bei Diagnosestellung ist der zukünftige Krankheitsverlauf, ob monophasisch oder multiphasisch, derzeit nicht zu prognostizieren. Aus diesem Grund sollte nach einem ersten Ereignis nur in Ausnahmefällen eine DMT eingeleitet werden und vorerst der weitere Krankheitsverlauf beobachtet werden.
MS und verwandte neuroimmunologische Erkrankungen stellen für behandelnde Neurolog:innen trotz großer jüngster Fortschritte nach wie vor eine Herausforderung dar. Für ein effektives Management bedarf es einer frühzeitigen korrekten Diagnose, um die Morbidität zu reduzieren und die Lebensqualität der Betroffenen zu verbessern.

Dr.in Franziska Di Pauli, PhD
Universitätsklinik für Neurologie, Medizinische Universität Innsbruck

Autor:
Univ.-Prof. Priv.-Doz. Dr. Christian Enzinger, MBA
Univ.-Prof. Priv.-Doz. Dr. Christian Enzinger, MBA
Präsident der Österreichischen Gesellschaft für Neurologie (ÖGN)

Ursprünglich erschienen:
AEK 09|2024
AEK 09|2024
Literatur bei den Verfasser:innen