






















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Arzneimittelstandards
Woher kommen sie, und was bedeuten sie für die Apotheke?
8. November 2024
Rechtlicher Hintergrund
Das Europäische Arzneibuch ist das einheitliche Referenzwerk für die Qualitätskontrolle von Arzneimitteln: Es ist in rund 40 Ländern rechtlich bindend, wird in über 100 Ländern angewandt und hat die Zielsetzung, Qualität und Sicherheit von Arzneimitteln in Europa zu gewährleisten. Was für Österreich mit den Regeln des Europäischen Arzneibuches nicht abgedeckt ist, wird durch die Vorschriften des Österreichischen Arzneibuches (ÖAB) geregelt – es stellt die nationale Ergänzung zum Europäischen Arzneibuch dar. Die in beiden Arzneibüchern enthaltenen Regeln bzw. Normen sind größtenteils Vorschriften zur Definition, Herstellung, Qualität, Zusammensetzung, Dosierung, Bezeichnung, Lagerung und Prüfung von Arzneimitteln sowie auch zu deren Verpackungen.
Arzneimittel müssen laut Arzneibuchgesetz nach diesen im Arzneibuch enthaltenen Regeln hergestellt, geprüft und in Verkehr gebracht werden; eine große Rolle in der (Apotheken-)Praxis spielen hier die Identitäts- und Reinheits- bzw. Qualitätsprüfungen. Hinsichtlich Vorschriften gibt es jedoch Unterschiede: Zur Prüfung dürfen auch andere Geräte verwendet bzw. Methoden angewendet werden, sofern sie dem Stand der Wissenschaft entsprechen und damit nachweislich die gleichen Ergebnisse wie mit der Methode des Arzneibuches erzielt werden. Bei der Herstellung hingegen dürfen nur dann andere Methoden angewendet werden, sofern eine Monografie des Arzneibuches ein anderes Herstellungsverfahren ausdrücklich erlaubt.
Dabei gelten die Vorschriften der jeweils gültigen Fassung: Eine neue Ausgabe des Europäischen Arzneibuches erscheint alle 3 Jahre, in der Zwischenzeit wird mit Nachträgen (sogenannten Supplements) gearbeitet. Vom Österreichischen Arzneibuch (ÖAB) gibt es jedes Jahr eine neue Ausgabe.
Wie entsteht eine Monografie im Arzneibuch?
Monografien bzw. Qualitätsstandards für das Europäische Arzneibuch entstehen durch das EDQM (European Directorate for the Quality of Medicines & HealthCare) mit Sitz in Straßburg (Frankreich) in einem umfassenden und strukturierten Prozess (Abb.).
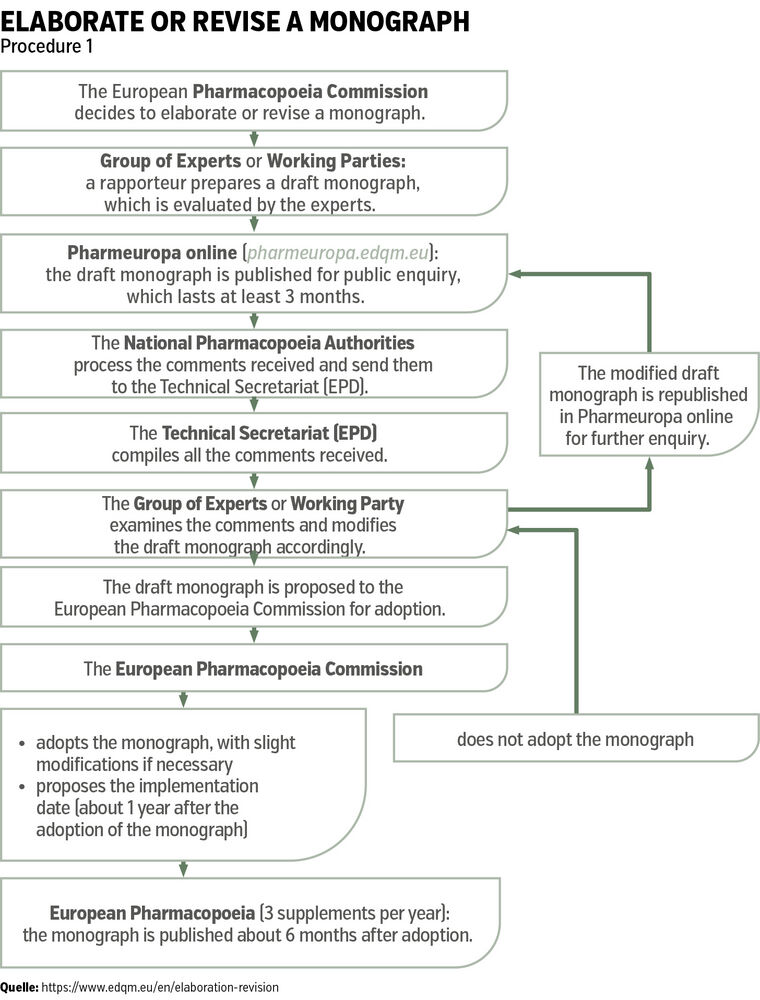
Vereinfacht gesagt läuft der Prozess folgendermaßen ab:
- Entsteht ein Bedarf an einer neuen oder aktualisierten Monografie für Arzneistoffe, Hilfsstoffe, Zubereitungen oder auch Methoden etc., so wird die Er- bzw. Überarbeitung durch die Europäische Arzneibuchkommission beschlossen. Vorschläge hinsichtlich eines Bedarfes bzw. einer Änderung können von nationalen Arzneimittelbehörden, pharmazeutischen Unternehmen, Wissenschafter:innen oder anderen Interessengruppen eingebracht werden.
- Nach dem Beschluss durch die Kommission wird nun in der jeweils thematisch passenden Expertengruppe ein Monografievorschlag erarbeitet und anschließend für mindestens 3 Monate auf der Plattform Pharmeuropa publiziert. Allfällige Kommentare zur vorgeschlagenen Monografie werden gesammelt und in der Expertengruppe behandelt, gegebenenfalls werden im Monografievorschlag nochmals Änderungen vorgenommen.
- Der finale Monografievorschlag wird der Europäischen Arzneibuchkommission zum Beschluss vorgelegt. Nach Beschluss wird die neue Monografie circa 6 Monate später im aktuellen Nachtrag (Supplement) veröffentlicht und tritt wiederum 6 Monate später in Kraft. Dies ermöglicht z. B. Firmen, sich auf die neuen verpflichtenden Standards einzustellen.
Die Erstellung einer neuen bzw. die Überarbeitung einer bestehenden Monografie für das Österreichische Arzneibuch läuft sehr ähnlich ab: Die Erarbeitung erfolgt durch eine Expertengruppe und die Beschlüsse durch die Kommission; auf Grund des geringeren Umfangs des Österreichischen Arzneibuches verglichen mit dem Europäischen Arzneibuch gibt es in Österreich nur eine Expertengruppe.
Die Rolle und Pflichten der Apotheke
Dank der hohen Qualitätsstandards und Qualitätskontrollen durch die herstellenden Betriebe (Rohstoff-, Arzneimittel- sowie auch Packmittelhersteller) fällt vieles an selbst durchzuführenden Kontrollen für die Apotheke bereits weg! Liegen daher – wie es meist der Fall ist – entsprechende Prüfzertifikate zur Qualitätsprüfung eines Arzneimittels bereits vor, so braucht die Apotheke die Prüfungen nicht durchzuführen; weiters bestünde auch die Möglichkeit, Qualitätsprüfungen an andere entsprechend befugte Betriebe auszulagern.
Anders verhält sich die Situation jedoch hinsichtlich Identitätsprüfung: Es ist gesetzlich vorgeschrieben, dass Arzneimittel (ausgenommen Arzneispezialitäten) vor Abgabe an den/die sogenannte:n Letztverbraucher:in immer auf Identität geprüft werden müssen! Das Arzneibuch berücksichtigt hier aber die Situation der Apotheken: Viele Monografien enthalten zwei Möglichkeiten zur Identitätsbestimmung (First bzw. Second Identification). Während für die Industrie die erste Identifikation (First Identification) vorgeschrieben ist, haben die Apotheken die Wahlmöglichkeit zwischen den beiden Varianten. Vorteil der zweiten Möglichkeit ist meist, dass dabei viele Geräte, wie zum Beispiel ein IR, nicht notwendig sind. In der Praxis kommen in der Variante der Second Identification meist simple Methoden wie die Bestimmung des Schmelzpunktes und einfache nasschemische Nachweise zur Anwendung.
Selbst wenn das Arzneibuch keine diesbezüglichen Angaben enthält, ist eine Identitätsprüfung dennoch verpflichtend: Hier orientiert man sich einfach am Stand der Wissenschaft, bzw. Hersteller leisten hier oft Hilfestellung – so werden einige Rohstoffe mittlerweile mit einem fertigen „Kit“ zur Identitätsbestimmung ausgeliefert. Die Durchführung der Identitätsprüfung muss dokumentiert werden, die Aufbewahrungsfrist dafür beträgt 5 Jahre.

Autorin:
Julia Mösslacher, PhD
Julia Mösslacher, PhD
Gastprofessorin für Pharmazeutische Technologie & Biopharmazie, Paracelsus Medizinische Privatuniversität

Ursprünglich erschienen:
Apo-K 21|2024
Apo-K 21|2024
Bildnachweis
Vorschaubild: © Piya-W - stock.adobe.com