





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Hoffnung Corona-Impfung
18. November 2020
ARZT & PRAXIS: Wie lange dauern üblicherweise die Erforschung und das entsprechende Zulassungsverfahren eines neuen Impfstoffes?
 Dr. Barbara Tucek: Die Dauer der Forschung und Entwicklung hängt immer davon ab, ob es nur ein weiterer Impfstoff gegen eine bekannte Krankheit ist oder ob es tatsächlich etwas ganz Neues ist wie jetzt bei COVID-19. Gibt es zum Beispiel bereits Impfstoffe gegen ein bestimmtes Virus, dann genügt es für die Zulassung, dass das Erreichen des sogenannten immunologischen Schutzkorrelats mittels durch Impfung induzierter Antikörperkonzentrationen im Blut des Impflings nachgewiesen wird: Wenn der Antikörper-Titer einen gewissen Wert erreicht, ist dies mit Schutz vor der jeweiligen Erkrankung gleichzusetzen. Ist jedoch kein Schutzkorrelat etabliert, sind große und aufwendige Phase-III-Studien erforderlich, um Wirksamkeit im Verhältnis zur Verträglichkeit nachzuweisen.
Dr. Barbara Tucek: Die Dauer der Forschung und Entwicklung hängt immer davon ab, ob es nur ein weiterer Impfstoff gegen eine bekannte Krankheit ist oder ob es tatsächlich etwas ganz Neues ist wie jetzt bei COVID-19. Gibt es zum Beispiel bereits Impfstoffe gegen ein bestimmtes Virus, dann genügt es für die Zulassung, dass das Erreichen des sogenannten immunologischen Schutzkorrelats mittels durch Impfung induzierter Antikörperkonzentrationen im Blut des Impflings nachgewiesen wird: Wenn der Antikörper-Titer einen gewissen Wert erreicht, ist dies mit Schutz vor der jeweiligen Erkrankung gleichzusetzen. Ist jedoch kein Schutzkorrelat etabliert, sind große und aufwendige Phase-III-Studien erforderlich, um Wirksamkeit im Verhältnis zur Verträglichkeit nachzuweisen.
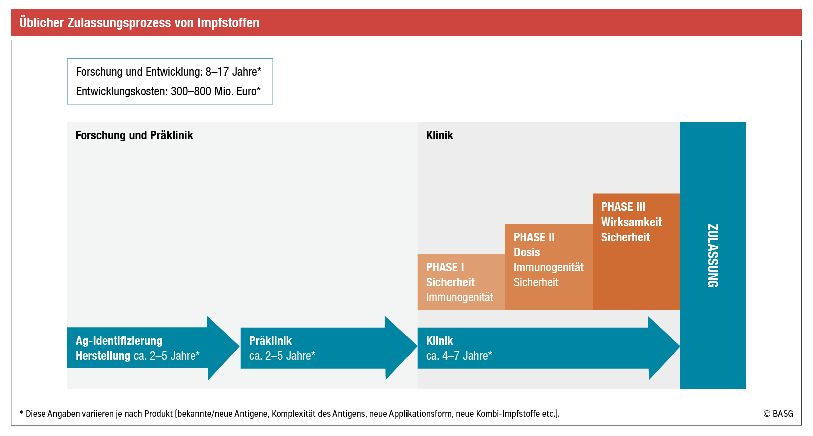
Die Dauer bis zur Marktreife ist von einer Vielzahl an Faktoren – Kenntnis über das jeweilige Krankheitsgeschehen, neues Antigen, neue Herstellungstechnologie etc. – abhängig, deswegen kann man nur einen Durchschnitt nennen. In der Regel dauert die Entwicklung eines neuen Impfstoffes etwa 10–15 Jahre, so z. B. bei HPV und Rotaviren; bei Varizellen und dem nasalen Influenza- Impfstoff waren es hingegen 25–30 Jahre. Gegen HIV oder Hepatitis C gibt es aber noch immer keine Impfung, obwohl bereits seit 30 Jahren daran geforscht wird. Eine Übersicht zu Zulassungsverfahren von Impfstoffen im Allgemeinen (Abb.) und zur Zulassung von COVID-19-Impfstoffen im Speziellen findet sich auf der AGES-Homepage.
Bei der Forschung zu einem SARS-CoV-2-Impfstoff wird davon gesprochen, dass man an die Forschungsergebnisse zu einem Impfstoff gegen SARS-CoV-1 anknüpfen kann, den es allerdings nicht gibt …
Die Forschung zu einem SARS-CoV-1-Impfstoff wurde eingestellt, da diese SARS-Epidemie glücklicherweise rasch wieder zum Erliegen gekommen ist. Wenn eine Krankheit nicht mehr existiert, ist die Erforschung eines Impfstoffes für die Pharmaindustrie natürlich nicht mehr attraktiv. Aber die Erfahrungen und Forschungsergebnisse zu SARS-CoV-1 und zu MERS, das im arabischen Raum vorkommt, konnte und kann man bei der Entwicklung eines neuen Corona-Impfstoffes natürlich nutzen und somit Entwicklungszeit sparen. Nicht wenige Firmen arbeiten gerade im Bereich der mRNA- oder der vektorbasierten Impfstoffe an sogenannten Plattformen, die grundsätzlich als Basis für die Entwicklung von Impfstoffen gegen viele Infektionskrankheiten zum Einsatz kommen könnten.
Bei vektorbasierten Impfstoffen, die auch zu den Lebendimpfstoffen zählen, bedient man sich eines an sich harmlosen Virus, wie z. B. eines attenuierten Adenovirus. Dabei werden in diese viralen Vektoren, die in ihrer Funktion nun einem trojanischen Pferd gleichen, Gene eingebaut, die die Information für SARS-CoV-2-Oberflächenproteine beinhalten, gegen die in weiterer Folge Antikörper gebildet werden sollen. Eine dieser unter anderem auch bereits für die Impfstoffforschung gegen MERS genutzten Plattformen wird jetzt für die SARS-CoV-2-Impfstoffentwicklung eingesetzt.
mRNA- und DNA-Impfstoffe sind derzeit beim Menschen noch nicht zugelassen, man hat sich aber mit der Technologie bereits über viele Jahre beschäftigt. Hier knüpft man jetzt an und kann dadurch auch in der Entwicklung abkürzen.
Es finden natürlich präklinische und klinische Studien mit den gegenständlichen Produkten statt, aber um zusätzlich Zeit einzusparen, wird nun das Entwicklungsprogramm kompakt gestaffelt, mit sich überlappenden Entwicklungsphasen. Und hier muss man betonen, dass das Risiko bei den forschenden Firmen liegt. Risiko deshalb, weil dadurch eventuell kein bestgeeigneter Impfstoffkandidat für die Phase-III-Studien erarbeitet wurde und möglicherweise das gesamte Projekt zu einem relativ späten Zeitpunkt noch vor Einreichung zur Zulassung scheitern könnte.
Reicht das, um den Entwicklungs- und Zulassungsprozess auf ein bis zwei Jahre zu verkürzen?
Grundsätzlich sind alle Versuche am Tier und am Menschen in Impfstoffstudien gemäß den rigorosen wissenschaftlich-regulatorischen Vorgaben auch für COVID-19-Impfstoffe durchzuführen. Solange nicht bekannt ist, welche Antikörperkonzentration mit Schutz gleichzusetzen ist, müssen nach erfolgreichem Abschluss der Tierversuche bzw. auch überlappend, Studien am Menschen mit Phase-I-, Phase-II- und den großen Phase-III-Studien zum Nachweis der Wirksamkeit und Sicherheit durchgeführt werden.
Normalerweise werden diese Schritte hintereinander durchgeführt, um basierend auf den jeweiligen Ergebnissen zu entscheiden, ob weiter in die sehr teure Entwicklung investiert werden soll oder nicht – jetzt findet vieles parallel statt. Gerade die Präklinik sowie die Phasen I und II sind wichtige Vorarbeiten, um die großen, aufwendigen und sehr teuren Phase-III-Studien durchzuführen, die ein positives Nutzen-Risiko- Verhältnis zeigen müssen, damit eine Zulassung ausgesprochen werden kann. Die Industrie hat daher auch ein größtmögliches Eigeninteresse daran, dass die ersten zwei Phasen gewissenhaft und korrekt durchgeführt werden, um nicht mit einem suboptimalen Impfstoffkandidaten weiterhin Zeit und Geld zu verlieren.
Auch wenn die Zulassungsverfahren verkürzt werden, wird es behördenseitig keine Abstriche – weder qualitativ noch im Umfang der Begutachtung – geben.
Wie wirksam muss ein Impfstoff überhaupt sein, um zugelassen zu werden?
Grundsätzlich muss der Nutzen mögliche Risiken (Nebenwirkungen) in der Zielpopulation überwiegen.
Dieses positive Nutzen-Risiko-Verhältnis an einem bestimmten Prozentsatz festzumachen, ist schwierig. Dafür gibt es zu viele Faktoren, die vom Impfling, vom Erreger und vom jeweiligen Impfstoff abhängen: z. B. Gefahrenpotenzial der vorzubeugenden Krankheit, Mutationen des Virus, Zielgruppe inkl. deren Immunkompetenz. Bei der Influenza liegt die Wirksamkeit in der Bevölkerung durchschnittlich bei 40–60 %1 bzw. auch darüber, wenn sich das Virus in der Saison nicht ändert. Diese Besonderheit unterscheidet die Influenzaimpfung von anderen Impfungen, die eine wesentliche höhere Wirksamkeit zeigen (z. B. Masern, FSME).
Welche sind die wichtigsten Nebenwirkungen, über die man aufklären muss?
Das hängt im Detail vom jeweiligen Impfstoff ab. Allgemeinreaktionen wie Müdigkeit, Kopfschmerzen und Fieber bzw. Lokalreaktionen mit Rötung, Schwellung und Schmerzen an der Einstichstelle zählen zu den häufigsten Impfreaktionen, die nach nahezu jeder Impfung möglich sind. Sehr selten kommen Impfkrankheiten vor wie Impfmasern, die allerdings in der Regel komplikationsfrei und nicht ansteckend sind, oder Arthralgien nach einer Röteln- Impfung. Echte Impfkomplikationen sind eine Rarität, in einer Größenordnung von 1:500.000–1:1.000.000 bei allergischen Reaktionen oder von 1:1,5 Millionen verabreichten Dosen beim anaphylaktischen Schock. Grundsätzlich gilt: Auch bei keinem einzigen anderen Arzneimittel können schwerere Nebenwirkungen mit absoluter Sicherheit ausgeschlossen werden.
Wichtig ist, zu erwähnen, dass die Arzneimittelbehörden das Produkt nach der Zulassung laufend überwachen, ab dann greift die Pharmakovigilanz (Arzneimittelüberwachung). Dabei werden alle erhobenen und gemeldeten vermuteten Nebenwirkungen, inkl. Erfahrungen mit besonderen Personengruppen, wie z. B. Schwangeren, erhoben. Des Weiteren findet auch ein internationaler Austausch mit den Zulassungsbehörden in der EU und auch der WHO statt.
Bei seltenen Erkrankungen ist eine Zulassung eines Arzneimittels auch nur mit Daten einer Phase-I/II-Studie möglich. Wäre das bei einer SARS-CoV- 2-Impfung auch denkbar?
Nein, COVID-19 ist natürlich auch keine seltene Erkrankung. Hier hat sich die EMA bereits geäußert: Solange so wenig über das Virus bekannt ist, solange man das immunologische Schutzkorrelat nicht kennt, solange braucht es Wirksamkeits- und Sicherheitsstudien. Das bedeutet, dass es in der EU ohne Phase- III-Studien derzeit keine Zulassung geben wird.
Die Grippeimpfung muss jedes Jahr neu gematcht werden. Braucht es dann auch jedes Jahr eine neue Zulassung?
Die Grippe zeichnet sich dadurch aus, dass – mit sehr seltenen Ausnahmen – jährlich neue Virusstämme kursieren. Daher ist eine jährliche Anpassung der saisonalen Influenza-Impfstoffe nötig. Die WHO empfiehlt jedes Jahr jeweils für die Nord- und die Südhalbkugel bestimmte Stämme. Eine komplette Neuzulassung Jahr für Jahr wäre allein schon zeitlich gesehen nicht möglich, ist aber auch nicht erforderlich. Früher gab es tatsächlich kleine klinische Prüfungen, bei denen Immunogenität und Sicherheit überprüft wurden. Nachdem sich dabei aber über die Jahre nie Abweichungen gezeigt haben, wurden keine Studien mehr durchgeführt. Das bereits zugelassene Produkt wird hinsichtlich der Influenzastämme abgeändert. Es wird allerdings vom Zulassungsinhaber ein Änderungsverfahren eingereicht, bei dem die jeweilige neue Zusammensetzung inkl. Herstellung von Behördenseite genau überprüft wird.
Alle Impfstoffe, so auch Grippeimpfstoffe, zählen zu den chargenfreigabepflichtigen Arzneimitteln. Das bedeutet, dass jede Charge einer Prüfung inkl. Freigabe durch ein behördliches Prüflabor (OMCL) unterliegt, bevor sie auf den Markt kommt.
Woran liegt es, dass gewisse Arzneimittel zwar in der EU zugelassen, in Österreich aber nicht verfügbar sind?
Teilweise geht es dabei tatsächlich um zu niedrige Produktionskapazitäten. Wie bereits erwähnt, gibt es über die Jahre gesehen immer weniger Impfstoffhersteller. Wenn nur eine einzige Firma einen bestimmten Impfstoff herstellt, kann es natürlich relativ leicht zu Lieferengpässen und im Extremfall sogar zu Versorgungsengpässen kommen. Die Ursachen können in einer Rohstoffverknappung, aber auch in einer plötzlich gesteigerten Marktnachfrage in diversen Ländern liegen (siehe Grippeimpfstoffe: hohe Nachfrage 2020). Impfstoffe zählen zu den biologischen Arzneimitteln, die einer sehr komplexen Herstellung unterliegen. Es kann bisweilen ein bis zwei Jahre dauern, um die Produktion entsprechend der Nachfrage anzukurbeln.
Ein weiterer Faktor können Preisverhandlungen und Abnahmegarantien sein. Die österreichische Bevölkerung war im internationalen Vergleich beispielsweise bei Influenza bisher nicht sonderlich impffreudig, und die Firmen müssen sich natürlich an der Durchimpfungsrate und an der Bevölkerungszahl orientieren, wenn sie in den Markt eintreten.
Viele der in Entwicklung bzw. in Studien befindlichen Corona-Impfstoffe funktionieren (z. B. als mRNA-Impfstoffe) nach einem neuen Prinzip. Gibt es bereits Erfahrungen mit diesen neuen Technologien?
Es ist bereits ein viraler Vektorimpfstoff gegen Ebola zugelassen. Es gibt auch Gentherapien, die nach diesem Prinzip funktionieren, um quasi Gen-Reparaturmechanismen in die Zelle zu schleusen. mRNA- oder DNA-Vakzine sind bisher noch nicht für den Menschen zugelassen, jedoch wird seit vielen Jahren daran geforscht.
Die in Entwicklung befindlichen SARS-CoV-2-Impfstoffe zielen prinzipiell auf ein immunogenes Oberflächenprotein des Virus ab, das SARS-CoV-2-Spikeprotein (oder Teile davon). Dieses Protein erzielt eine gute Immunantwort und es gibt unterschiedliche Wege, um dieses Protein dem Immunsystem als Antigen zu präsentieren. Klassisch wäre, das Virus zu verwenden und daraus entweder in abgeschwächter Form einen Lebendimpfstoff oder nach Inaktivierung durch Formaldehyd oder Hitze einen Totimpfstoff zu produzieren. Eine weitere bekannte Technologie sind die proteinbasierten Impfstoffe, wo nur noch einzelne Proteine, die die Immunantwort auslösen, verabreicht werden (Subunit-und Virus-like particles, z. B. als Grippeimpfstoffe), die aber sehr aufwendig in der Herstellung sind.
Welche SARS-CoV-2-Impfstoffe sind in den Zulassungsverfahren am weitesten fortgeschritten?
Am weitesten fortgeschritten sind vektorbasierte, mRNA-, proteinbasierte und herkömmliche Totimpfstoffe.
Was sind die Vorteile der neuen Technologien?
Wenn man an Viren forscht, um einen Lebend- oder Totimpfstoff herzustellen, muss in Hochsicherheitslabors gearbeitet werden, da man es mit potenziell gefährlichen Viren(mengen) zu tun hat. Die Herstellung ist aufwendig und langwierig. Bei einem mRNA-Impfstoff benötigt man kein solches Hochrisikolabor – es handelt sich um einen ganz anderen Herstellungsprozess, bei dem die mRNA biotechnologisch hergestellt wird. Zudem verläuft dieser Prozess ungleich schneller: Eine Impfstoffcharge ist binnen 1–2 Wochen fertig produziert. Es können sehr große Mengen hergestellt werden, und das ist vor allem in Pandemiezeiten ein wichtiges Thema, weil man sehr schnell, exakt und in großer Menge mRNA-Vakzine verfügbar machen kann. Die eigentliche Impfstoffproduktion wird in die körpereigene Zelle verlagert, wenn man so will. Die eingeschleuste mRNA veranlasst Zellorganellen, die gewünschten Proteine zu erzeugen, die eine Immunantwort auslösen sollen.
Ist die Sorge vieler Menschen berechtigt, die einen Eingriff in unser Erbgut befürchten?
Der Ablauf in der Zelle, der durch die Zuführung der mRNA initiiert wird, ist nichts Außergewöhnliches für unseren Körper. Dieser Prozess, Proteine gemäß spezifischer genetischer Information der mRNA herzustellen, ist etwas, das laufend in uns geschieht. Zudem ist mRNA extrem instabil, verbleibt nicht lange in unserem Körper und kann nicht in das Erbgut integrieren. Die Herausforderung liegt eher darin, dafür zu sorgen, dass das entsprechende Protein in ausreichender Menge erzeugt wird, um eine optimale Immunantwort hervorrufen zu können.
DNA-Impfstoffe gibt es bislang nur im Veterinärbereich. Hier wird beispielsweise mittels Elektroporation die DNA in den Zellkern verbracht. Dabei wird die Ladung an der Zellmembran so verändert, dass kleine Löcher – Poren – entstehen und die DNA nicht nur in die Zelle, sondern auch in den Zellkern verfrachtet wird.
Die gerade für Europa sehr wichtige Phase-III-Studie zu dem Impfstoff von AstraZeneca musste unterbrochen werden …
Ja, dazu muss allerdings gesagt werden, dass ganz allgemein Unterbrechungen von Studien aufgrund von vermuteten Sicherheitsproblemen laufend stattfinden. Klinische Studien werden häufig unterbrochen, wenn fragliche Komplikationen auftreten. Dies ist der Probandensicherheit geschuldet und entspricht den strikten Regularien der klinischen Prüfung.
In diesem Fall gab es jedoch eine große mediale Beachtung. Es zeigt vor allem, dass die vorgegebenen Maßnahmen selbstverständlich auch hier greifen, trotz des enormen Zeitdrucks. Ein unabhängiges Expertenkomitee bewertet den Fall und prüft, ob es einen Kausalzusammenhang zwischen der vermeintlichen Komplikation und der Impfung gibt oder nicht. Es gibt gemäß den Vorgaben grundsätzlich zwei Gründe, Studien abzubrechen: Das Risiko überwiegt den Nutzen oder das Produkt ist so wirksam, dass es unethisch wäre, dieses Arzneimittel der Zielpopulation noch länger vorzuenthalten.
Würden Sie sich persönlich gegen SARS-CoV-2 impfen lassen, sobald eine Impfung verfügbar ist?
Ja, unter zwei Voraussetzungen: Erstens muss es ein EU-weit zugelassener Impfstoff sein. Wenn er zugelassen ist, würde ich abwarten, für wen das Nationale Impfgremium den jeweiligen Impfstoff empfiehlt. Denn es kann ja sein, dass ein zugelassener Impfstoff nur für gewisse Risikogruppen empfohlen wird. Bei einer Empfehlung für eine Personengruppe, der auch ich angehöre, werde ich mich selbstverständlich impfen lassen.
Was halten Sie von einer Impfpflicht?
Hier kann ich nur für mich persönlich sprechen: Es muss immer versucht werden, wissenschaftlich gesicherte Information bestmöglich verständlich zu machen. Das ist etwas, das sicherlich noch nicht ausgeschöpft ist. Diese Informationsmaßnahmen müssen verbessert werden. Für gewisse Berufsgruppen, wie z. B. das Personal im Gesundheitswesen, gilt es neben dem Eigenschutz auch Verantwortung für die anvertrauten Schutzbedürftigen zu übernehmen. Ich verstehe persönlich nicht, dass es Menschen in diesen sozialen Berufen zu geben scheint, die dieses Selbstverständnis nicht mitbringen.
Die Wirksamkeit eines Impfstoffes zu vermitteln oder für den Laien verständlich nachzuweisen, ist oft schwierig. Bei anderen Medikamenten ist es simpel: Ein Patient ist krank, nimmt ein Arzneimittel und wird gesund. Bei der Impfung ist der Patient vorab zumeist gesund, bleibt gesund und bemerkt oft gar nicht, dass er das seinem durch Impfung „trainierten“ Immunsystem zu verdanken hat. Es sind daher auch immer weniger Firmen, die Impfstoffe produzieren, die das Know-how und die Durchhaltekraft in der aufwendigen Entwicklung haben und über die enormen finanziellen Mittel verfügen – von zunehmender Impfskepsis in den Industrieländern einmal abgesehen.
Bei COVID-19 ist man sich nun des Wertes eines verfügbaren, wirksamen und sicheren Impfstoffes plötzlich bewusst. Es sind jetzt alle erstaunt, wie rasch Wissen und Forschung vorangetrieben werden können und wie viel investiert wird.
Wenn aber alle Möglichkeiten der Motivation und Information ausgeschöpft sind und es trotzdem zu einem hohen Anstieg der Fallzahlen einer für die Volksgesundheit bedrohlichen Krankheit kommt, die durch eine Impfung verhinderbar wäre, denke ich persönlich hin und wieder schon über eine Impfpflicht nach. Gleichzeitig bin ich mir aber darüber im Klaren, dass Druck Gegendruck erzeugt und eine Impfpflicht kontraproduktiv sein könnte, weil dadurch radikale Impfgegner mehr Auftrieb erhalten und ihnen damit auch mehr Gehör und Zulauf verschafft werden könnten. Ich bin hier also letztlich (noch) unschlüssig.
Vielen Dank für das Gespräch!

Ursprünglich erschienen:
A&P 09|2020
A&P 09|2020