





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
ARZT & PRAXIS: Wie häufig ist Epilepsie bei Kindern?
OÄ Priv.-Doz.in Dr.in Gudrun Gröppel: Durchschnittlich erkranken etwa 40–50 von 100.000 Kindern an Epilepsie. Im ersten Lebensjahr ist die Anzahl sogar dreimal so hoch. Betrachtet man die Anfallsereignisse unabhängig von ihrer Ursache, so erleiden 4–10 % aller Kinder und Jugendlichen im Laufe ihres Lebens einen Anfall und bedürfen einer klinischen Abklärung.

Wie äußert sich Epilepsie bei Kindern am häufigsten?
Auch im Kindesalter gibt es verschiedene Arten von Anfällen und Anfallssyndromen. Es gibt generalisierte Epilepsien wie die juvenile Myoklonusepilepsie mit Zuckungen am ganzen Körper, aber auch fokale Anfälle, die von einem ganz umschriebenen Areal ausgehen und nur einzelne Gliedmaßen betreffen. Bei diesen Anfällen hängt die Symptomatik von der Lokalisation der epileptischen Aktivität im Gehirn ab.
Welche Erste-Hilfe-Maßnahmen sind sinnvoll und welche nicht?
Als erste Maßnahme sollte der Patient oder die Patientin in die stabile Seitenlage und aus einer etwaigen Gefahrenzone gebracht werden. Die Dauer des Anfalls muss unbedingt dokumentiert werden, da bei einem großen Anfall nach drei Minuten eine Notfallmedikation verabreicht werden muss. Wegen der hohen Verletzungsgefahr darf auf keinen Fall in den Mund gegriffen werden, ein Beißkeil oder den/die Patient:in festzuhalten ist obsolet.
Machen bereits Fieberkrämpfe eine antikonvulsive Therapie erforderlich?
Bei Fieberkrämpfen unterscheidet man zwischen einfachen und komplizierten Fieberkrämpfen. Einfache Fieberkrämpfe erfordern in der Regel keine Medikation, außer sie treten zu häufig auf. Komplizierte Fieberkrämpfe müssen immer dahingehend abgeklärt werden, ob nicht eine andere Erkrankung oder eine andere Epilepsieform dahinterliegt.
Gibt es Epilepsieformen, die häufig unerkannt bleiben bzw. besonders schwierig zu diagnostizieren sind?
Nächtliche Anfälle sind schwer zu diagnostizieren, da die Kinder im Normalfall alleine schlafen. Aber auch Epilepsieformen im Säuglingsbereich wie das West-Syndrom sind oft eine Herausforderung. Besonders schwierig ist die Diagnose eines bioelektrischen Status im Schlaf: Dabei hat das Kind keine oder kaum klinische Anfälle; sobald es einschläft, kommt es aber zu einer deutlich erhöhten zerebralen Erregungsbereitschaft. Diese Kinder werden oft nur durch eine Entwicklungsverzögerung auffällig. Deshalb ist es wichtig, auch bei diesen Kindern zumindest einmal ein EEG durchzuführen.
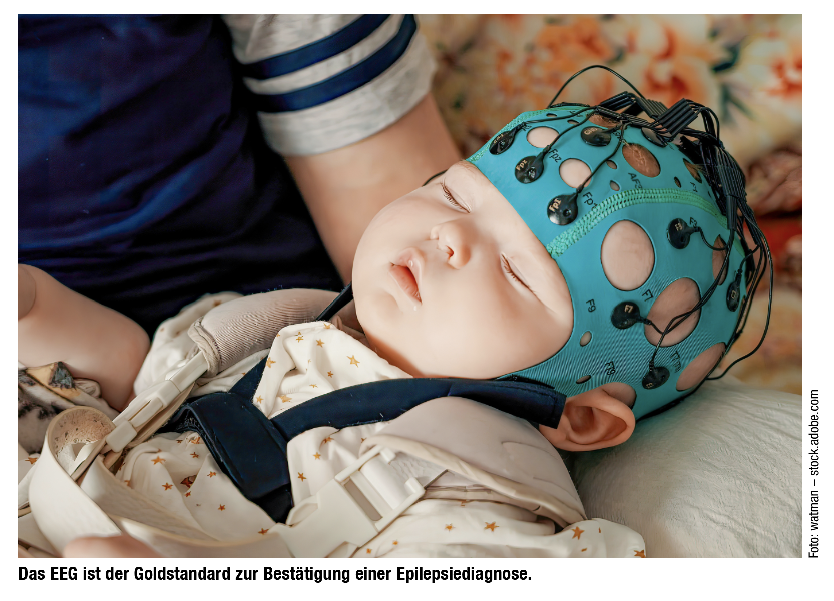
Wie kann zwischen epileptischen Anfällen und anderen neurologischen Störungen unterschieden werden?
Der Goldstandard ist das EEG, manchmal muss man aber auch ein prolongiertes Video-EEG-Monitoring durchführen, um etwaige Episoden direkt aufzunehmen. Hilfreich ist auch die Dokumentation der Anfälle durch ein Handyvideo durch die Eltern.
In welchen Fällen bedarf es unbedingt einer kinderneurologischen Abklärung?
Alle Kinder mit neurologischen Auffälligkeiten gehören primär von einem oder einer erfahrenen Kinderfacharzt/-ärztin begutachtet, der/die dann gegebenenfalls an eine Spezialabteilung überweist.
Sie leiten eine Ambulanz für schwer therapierbare Epilepsien. Wann gilt eine Epilepsie als „schwer therapierbar“?
Wenn zwei oder mehr anfallssupprimierende Medikamente nicht den gewünschten Erfolg, also Anfallsfreiheit, bringen, gilt diese Epilepsie als schwer therapierbar oder therapierefraktär. Dann muss man sich alternative Methoden überlegen.
Inwieweit hat die Epilepsie Einfluss auf die Kindesentwicklung?
Es gibt das Phänomen der „epileptischen Enzephalopathie“. Hier kommt es durch dauerhafte epileptische Entladungen im sich in Entwicklung befindlichen Gehirn zu einer Entwicklungsverzögerung. Prinzipiell ist dies nur durch rechtzeitige und adäquate Behandlung der Epilepsie zu vermeiden bzw. zu verbessern.
Wie wirkt sich die Therapie auf die geistige Entwicklung aus?
Natürlich ist die antikonvulsive Therapie mit Nebenwirkungen verbunden, darunter auch Veränderungen der Kognition oder des Verhaltens. Es ist aber wichtig zu betonen, dass keine Therapie so schwere kognitive Einbußen bewirken kann wie eine unbehandelte Epilepsie in Form der epileptischen Enzephalopathie.
Kann man bei Kindern mit Epilepsie eine Intelligenzminderung beobachten?
Je länger eine Epilepsie unbehandelt bleibt und je pathologischer das EEG ist, desto schwieriger wird es, eine altersentsprechende Entwicklung zu erreichen. Daher ist es wichtig, rasch mit einer Therapie zu beginnen. Es ist aber auch dann noch sinnvoll, therapeutisch einzugreifen, wenn die Anfälle schon länger bestehen, denn nur so kann ein Fortschreiten der Hirnschädigung verhindert werden. Zusätzlich besteht bei Kindern mit Epilepsie oft auch eine andere Grunderkrankung wie z. B. eine genetische Erkrankung, die per se schon mit einer Entwicklungsverzögerung einhergeht. Dementsprechend wichtig ist hier die Behandlung der Anfälle.
In welchen Fällen gelangt eine ketogene Diät (KD) zum Einsatz? Wann sind chirurgische Verfahren sinnvoll?
Die KD ist wie eine Medikation zu werten und auch so einzusetzen. Das Haupteinsatzgebiet ist die Säuglingsepileptologie, aber auch bei gewissen Stoffwechselerkrankungen ist die KD Mittel der ersten Wahl. Dazu zählt das GLUT-1-Defizit-Syndrom, bei dem das Gehirn Glukose nicht verarbeiten kann. Dann wird das Keton als Energielieferant angeboten und das Kind ist geheilt – ohne zusätzliche Therapie. Eine prächirurgische Evaluierung wird immer dann durchgeführt, wenn medikamentöse Maßnahmen nicht ausreichen oder nicht indiziert sind.
Welche sind die größten Hürden beim Management der kindlichen Epilepsie?
Das größte Problem besteht darin, dass die Anfälle nicht erkannt werden, wie es oft bei Blitz-Nick-Salaam(BNS)-Anfällen vorkommt, die in der Regel sehr kurz sind und relativ subtil sein können. Leider passiert es dann häufig, dass auch bei einer ersichtlichen Entwicklungsverzögerung den Eltern zum Zuwarten geraten wird. Ein weiteres Problem ist die Angst der Eltern vor der Medikation. Hier hilft nur eine konsequente Aufklärung über die Risiken der Anfälle bis hin zum „plötzlichen Tod von Epilepsiepatienten“ (SUDEP), aber auch über die Nebenwirkungen der Medikation. Bei therapierefraktärer Epilepsie wird oft zugewartet und Anfälle werden akzeptiert, anstatt an Epilepsiechirurgie zu denken.
Was wäre Ihre Botschaft an Kolleg:innen im niedergelassenen Bereich, die mit Epilepsie-Verdachtsfällen konfrontiert sind?
Gerade bei leichten oder unklaren Symptomen ist die Hemmschwelle der Kolleg:innen hoch, sofort an ein Universitätsklinikum zu überweisen. Insbesondere auch, da in den Spezialambulanzen die Wartezeit lang ist. Aber auch bei der Epilepsie gilt: „Time is Brain.“ Ich kann nur an die Kolleg:innen appellieren, bei einem Epilepsieverdacht mit Expert:innen in Kontakt zu treten, und das möglichst rasch. Gewisse Fragen kann man auch telefonisch klären, in manchen Fällen muss dann das Kind in die entsprechende Ambulanz geschickt werden.
Vielen Dank für das Gespräch!



Interview mit:
OÄ Priv.-Doz.in Dr.in Gudrun Gröppel
OÄ Priv.-Doz.in Dr.in Gudrun Gröppel
Leiterin der Neuropädiatrie und der pädiatrischen Funktionsdiagnostik an der Univ.-Klinik für Kinder- und Jugendheilkunde am Kepler Universitätsklinikum in Linz und Vorstandsmitglied der Österreichischen Gesellschaft für Epileptologie

Das Interview führte:
Dr. Sebastian Pokorny
Dr. Sebastian Pokorny

Ursprünglich erschienen:
A&P 08-09|2023
A&P 08-09|2023
Publikationsdatum: 2023-12-01
Zur Ausgabe »
Zur Ausgabe »