





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Alzheimer-Demenz und Glukosestoffwechsel: „Typ-3-Diabetes“?
27. November 2020
In Österreich leiden derzeit ca. 600.000 Personen an Diabetes mellitus und ca. 130.000 Personen an Demenz. 2050 werden doppelt so viele dement sein. 60–80 % der Demenzen entfallen auf die Alzheimer-Krankheit (AK), 15–20 % auf die vaskulären Demenzen, gefolgt von Lewy-Body-Demenzen mit 7–20 %. Die Prävalenzraten beider Erkrankungen sind deutlich gestiegen, und es besteht ein Zusammenhang.
Epidemiologische Evidenz
Die Rotterdam-Studie aus dem Jahr 1996 war eine der ersten, die ein erhöhtes Demenzrisiko bei Typ-2-Diabetes (T2D) dokumentierte. 2004 folgte die Religious Orders Study, die eine 65%-ige Steigerung des Demenzrisikos für T2D-Patienten gegenüber Kontrollpersonen ohne T2D belegte. Die Cache County Study 2006 zeigte ein erhöhtes Demenzrisiko bei T2D v. a. bei Frauen, und in der Framingham-Studie 2006 wurde für Subgruppen wie Patienten unter 75 Jahren mit T2D und Nichtträger des ApoEe4-Genotyps eine Risikoerhöhung für Demenz dokumentiert. In der Langzeitstudie des Kungsholmen Project aus Stockholm 2010 führten ein Prädiabetes und T2D zu einer um 3,18 Jahren kürzeren Progression von MCI (Mild Cognitive Impairment) zur Demenz. Die Vantaa-85+-Studie aus Finnland 2010 und die Hisayama-Studie 2011 fanden eine Verdoppelung des Demenzrisikos bei T2D-Patienten. Der Zusammenhang zwischen T2D und Demenz ist umso stärker, je früher diabetesbedingte Veränderungen auftreten.
T2D und die Alzheimer-Pathologie
Die Alzheimer- und Medikamentenforschung befasst sich seit drei Jahrzehnten mit Beta-Amyloid und Tau-Protein, und viele Forscher glauben, damit in einer Sackgasse zu sein. Neuritische Plaques (Beta-Amyloid) und hyperphosphoryliertes Tau-Protein (Neurofibrillen, Tangles) sind pathognomonisch für die AK. Ob diese pathologischen Proteine ursächlich sind und am Anfang der Erkrankung stehen, ist bis heute nicht wissenschaftlich überzeugend dokumentiert. Deshalb sind alternative Erklärungsansätze wie z. B. die Neuroinflammationshypothese oder der zerebrale Glukose- und Insulin-Stoffwechsel (STW) wieder ins Zentrum der Forschung gerückt. Seit den 1980er-Jahren ist aus PET-Untersuchungen bekannt, dass Patienten mit MCI bereits eine Reduktion des zerebralen Glukose-Stoffwechsels aufweisen. In einer kontrollierten Studie wurden bei kognitiv normalen älteren Patienten mit einer Glukoseintoleranz und bei Patienten mit einem neu diagnostizierten T2D spezielle Gehirnregionen mit F18-markierter Fluordesoxyglukose untersucht. Die Aktivität im parieto-temporalen, frontalen und cingulären Kortex, die als Risikoregionen für Alzheimer gelten, war während der Gedächtnistests sowohl bei Glukoseintoleranz als auch bei Diabetes im Gegensatz zu Gesunden deutlich reduziert. Das Muster entsprach den Veränderungen, die auch bei MCI und Alzheimer-Demenz (AD) dokumentiert sind. Eine Folge des zentralen Energiemangels sind Sekundärveränderungen wie neuritische Plaques und Tangles. Ein chronischer Stress-STW mit gesteigerten Cortisol-, Noradrenalin- und Ammoniakspiegeln könnte über Jahre hinweg die Insulinrezeptoren und den Insulin-STW beeinträchtigt haben. Eine reduzierte zentrale Insulinkonzentration oder -sensitivität führte zu besonders hoher Amyloidplaque-Bildung in glukosearmen Arealen, wie spezielle Amyloid-PET-Untersuchungen zeigten.
Die STW-Störung dürfte der toxischen Amyloidbildung vorausgehen.
Potenziell unterschiedliche Mechanismen können bei T2D das Demenzrisiko erhöhen; vor allem vaskuläre Veränderungen, kardiovaskuläre Erkrankungen, eine verminderte zerebrale Perfusion, „small vessel disease“ und zerebrale Insulte erhöhen das Demenzrisiko. Periphere Insulinresistenz wurde mit geringerer Glukoseaufnahme, reduzierter zerebraler Perfusion und Hirnatrophie in Zusammenhang gebracht. Eine zentrale Insulinresistenz (charakterisiert durch Down- Regulierung von Insulinrezeptoren), geringere Insulinbindung und abnorme Insulinsignale sind spezielle Merkmale des Alzheimer- Gehirns. Aus Tierversuchen wissen wir, dass T2D die Alzheimer- Pathologie mit Amyloid-Plaques, die Tau-Pathologie und die Neurodegeneration beschleunigen kann.
Eine jahrelange Behandlung mit Antidiabetika kann die AD-Pathologie abschwächen. Die kombinierte Behandlung mit Insulin und oralen Antidiabetika führte zu signifikant weniger Amyloid-Plaques in typischen Alzheimer-Regionen.
Alzheimer-Demenz als Diabetes Typ 3?
In manchen Forschungskreisen wird die AD auch als „Typ-3-Diabetes“ bezeichnet, da Patienten mit einem chronisch erhöhten Blutzucker (BZ) oftmals die Spätform der AD entwickeln. Suzanne de la Monte aus den USA prägte diesen Begriff erstmals vor 15 Jahren. Die Verbindung von Diabetes und Alzheimer beruht nicht auf Insulinproblemen per se, sondern auch auf dem Insulin-degrading Enzyme (IDE), das Insulin im Blut und Beta-Amyloid-Proteine im Gehirn abbaut. Wenn das Ziel von IDE v. a. Insulin ist und die Insulin-Spiegel die IDE-Produktion übertreffen, so wird die Fähigkeit von IDE, amyloidogene Proteine abzubauen, deutlich reduziert sein, und in der Folge werden mehr zerebrale Plaques mit Beta-Amyloid entstehen. Dieses Phänomen kann auch Personen mit Prädiabetes mit einer Glukoseintoleranz und chronischen Hyperglykämien betreffen. Umgekehrt kann die konsequente BZ-Kontrolle präventiv gegen funktionellen zerebralen Abbau wirken. Der Prävalenzanstieg von M. Alzheimer und Diabetes ist teilweise durch falsche Ernährung mit chronischen Hyperglykämien erklärbar, wodurch IDE darin gehemmt wird, Insulin und Beta-Amyloid abzubauen.
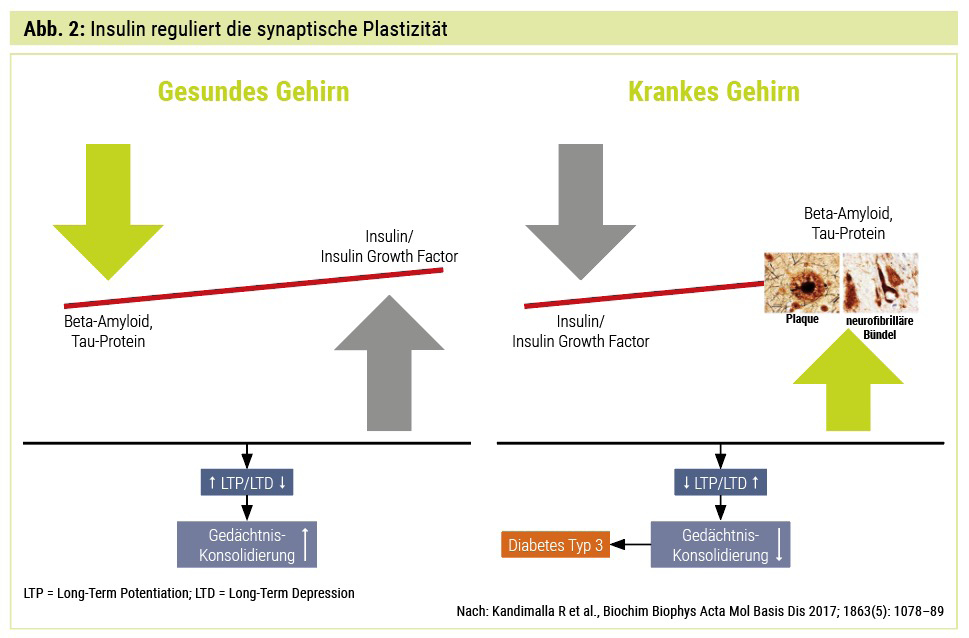
Es besteht eine hohe epidemiologische Evidenz, dass T2D – wegen des Versagens der Glukoseabsorption in Neuronen zur Energie- Produktion – eindeutig mit kognitiven Minderleistungen korreliert. Die Beziehung zwischen T2D und AD ist sehr komplex und verbunden mit Insulinresistenz, Insulin-like-Growth-Factor-(IGF-)Signalgebung, Insulin-degrading-Enzyme-(IDE-)Hemmung, oxidativem Stress, Inflammationsreaktion, Glykogensynthasekinase-3β-(GSK3β-)Signalmechanismen, Amyloid-Precursor-Protein-Umwandlung zum pathologischen Amyloid-beta (Aβ), neurofibrilläre Tangle-Entstehung und Regulierung der zentralen Acetylcholinesterase-Aktivität (Abb. 1 und 2). Wegen dieser gemeinsamen pathophysiologischen zellulären und molekularen Wegstrecken zwischen T1D, T2D und AD benannten Forscher diese als Diabetes Typ 3.
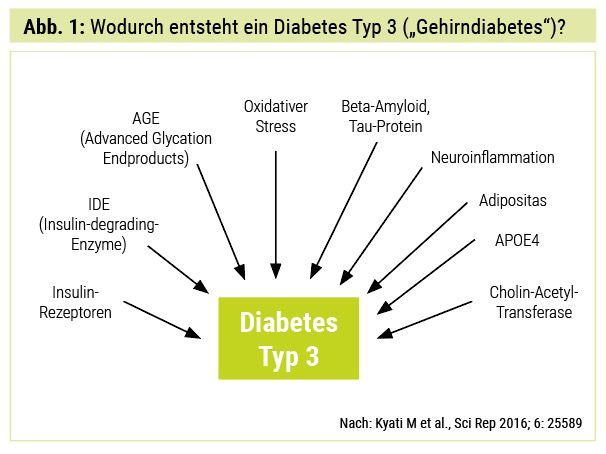
Beeinflussen Antidiabetika das Demenzrisiko?
Durch den Mangel an effizienten Behandlungsmöglichkeiten für die AK verlagerte sich der Fokus auf die Identifizierung von möglichen modifizierbaren Risikofaktoren (RF). Diese sind kardiovaskuläre Erkrankungen und Lebensstilfaktoren wie Ernährung und physische Aktivität. Die hohe Evidenz, dass T2D das Demenzrisiko signifikant steigert, führte zur Frage, ob Antidiabetika präventiv wirken und das Demenzrisiko reduzieren können. Interessant ist, dass gehirngängige Statine, wie Atorvastatin oder Simvastatin, in epidemiologischen Studien eine reduzierte Alzheimer-Rate erzielen konnten. Statine bewirken, dass Mikrogliazellen vermehrt IDE ausscheiden. IDE baut nicht nur Insulin, sondern auch Beta-Amyloid ab. IDE-Konzentrationen waren in Prädilektionsstellen für AD, wie z. B. im Hippocampus, deutlich reduziert. Möglicherweise ist dies die Folge einer Insulinresistenz oder Herunterregulation des Enzyms IDE.
Nur wenige Antidiabetika wurden in Beobachtungsstudien als Mono- oder Kombinationstherapien in Bezug auf das Demenzrisiko erforscht, noch weniger in klinischen Studien getestet.
Metformin wird zumeist als erstes Antidiabetikum verordnet und führt zu reduzierter Glukoseproduktion in der Leber, vermehrter peripherer Glukoseaufnahme und verbesserter Insulinsensitivität. In einer großen Gesundheitsdatenbank aus Taiwan mit 25.393 Patienten mit Diabetes war das Demenzrisiko im Jahr 2011 um das 2,6-Fache erhöht, und die Metformin-Behandlung konnte das Demenzrisiko um 24 % senken. Sulfonylharnstoffe reduzierten das Demenzrisiko um 15 %. Die kombinierte Behandlung reduzierte das Demenzrisiko über 8 Jahre um 35 %. In einer deutschen Langzeitstudie aus dem Jahr 2015 an insgesamt 145.928 Patienten ohne Demenz war die Nichtbehandlung mit Pioglitazonen mit einem 23%-ig erhöhten Demenzrisiko verbunden. Hingegen senkte die Langzeitbehandlung mit Pioglitazon die Demenzinzidenz.
Allerdings weisen nicht alle Antidiabetika-Studien eine demenzprotektive Wirkung auf. In einer Studie war die Metformin-Behandlung mit erhöhtem AD-Risiko assoziiert, und in der Rotterdam-Studie aus dem Jahr 1996 war die Demenzinzidenz am höchsten bei mit Insulin Behandelten.
Sind Antidiabetika neue Antidementiva?
Zahlreiche Studien wiesen eine günstige Wirkung von Antidiabetika auf die Alzheimer-typische Gehirnpathologie nach, wie Amyloid- Plaques, Tau-Pathologie, Synapsenverlust, oxidativer Stress, Neurogenese, Neuroinflammation und kognitive Funktionen. In klinischen Studien am Menschen waren Antidiabetika leider nur in kleinen Studien erfolgreich. Metformin zeigte bei 80 Patienten mit amnestischen MCI positive Effekte auf das verbale Gedächtnis, und in einer anderen Studie bei Personen ohne Diabetes auf die exekutiven Funktionen.
Der Insulin-Sensitizer Rosiglitazon konnte in einer kleinen randomisierten Pilotstudie bei Patienten mit AD oder amnestischen MCI über 6 Monate sowohl die verzögerte Erinnerung als auch die Aufmerksamkeit verbessern. In einer offenen Studie an 42 Patienten wurde unter Pioglitazon in der Alzheimer Disease Assessment Scale (ADAS-Cog) eine Verbesserung dokumentiert. In größeren randomisierten klinischen Studien bei 518 Patienten mit leichter bis mäßiger AD konnte Rosiglitazon weder in der 2- oder 4- noch in der 8-mg-Dosierung gegenüber Placebo eine signifikante Überlegenheit nach 24 Wochen nachweisen. Auch eine Follow-up-Studie an 693 Patienten, die nach ihrem APOE-Genotyp stratifiziert wurden, zeigte keine signifikanten positiven Rosiglitazon-Wirkungen. Die größte Studie mit Pioglitazon 0,8 mg bei 25.000 AK-Patienten, die nach ihrem APOE-und TOMM40-Genotyp gescreent wurden, wurde vorzeitig wegen Nichtverzögerung der MCI-Entwicklung abgebrochen.
Wie wichtig Insulin für unsere Hirnleistung ist, zeigt sich durch Studien an Gesunden: Intranasal verabreichtes Insulin führte zu kognitiven Leistungssteigerungen. Besonders interessant waren Forschungen, dass in Zellkulturen Insulin die Beta-Amyloid-Bildung reduzieren konnte. Dieser Effekt konnte durch die Zugabe von Glitazonen gesteigert werden. In einer ersten Pilotstudie mit 24 Patienten mit MCI oder milder AD im Jahr 2008 verbesserten sich durch intranasales Insulin das verbale Gedächtnis und die Aufmerksamkeit. In einer anderen Studie zeigten nur ApoE4-Nichtträger Gedächtnissteigerungen, während ApoE4-Träger einen Gedächtnisabfall aufwiesen. 2013 wurde die SNIFF-Studie (Study of Nasal Insulin in the Fight against Forgetfulness) durchgeführt. 289 Patienten mit MCI oder AD wurden an 26 Zentren eingeschlossen. Intranasales Insulin war nicht wirksam in den kognitiven Leistungstests; allerdings musste der Applikator wegen Dysfunktionalität in der Mitte der Studie ausgewechselt werden. In dieser Studie war kein Effekt auf Liquor-Biomarker nachweisbar. Dieselbe Forschergruppe dokumentierte allerdings 2017 eine Reduktion von Phospho-Tau/Aβ42 durch Insulin.
In der ACCORD-Studie wurde eine besonders strenge Glykämie- Kontrolle bei T2D-Patienten mit einem HbA1c < 6 % durchgeführt. Die besonders intensiv behandelte Gruppe musste wegen erhöhter Mortalität sogar gestoppt werden. 47 Monate nach Beginn war kein positiver Behandlungseffekt auf die kognitiven Leistungen dokumentiert. Hypoglykämien trugen zu kognitiven Verschlechterungen bei.
Warum gibt es noch immer keinen Durchbruch in der Alzheimer-Therapie?
Wahrscheinlich ist der Zeitpunkt einer noch so vielversprechenden Intervention bereits zu spät, da Patienten mit MCI oder leichter Demenz bereits eine hohe Plaque- und Neurofibrillen-Dichte aufweisen und Synapsenverluste und Neurodegeneration vorliegen. Möglicherweise wären Insulinsensitizer bei bestätigter zentraler oder peripherer Insulinresistenz erfolgreicher. ApoE4 trägt zur metabolischen Dysregulation bei und ist deshalb eine besonders kritische Variable für den Therapieerfolg unter Antidiabetika. Die Studienlage, ob ApoE4-Carrier oder -Nicht-Carrier besser ansprechen, ist inkonsistent. Es existieren Hinweise, dass eine T2D-Kombinationstherapie einer Monotherapie überlegen sein dürfte. Ob T2D das Gehirn direkt über einen insulinvermittelten Mechanismus oder durch Begleiterkrankungen (wie Dyslipidämie, Adipositas, Hypertonie) oder über AGEs (advanced glycation end products) schädigt, ist nicht restlos geklärt. Deshalb wird die Therapie der Zukunft Antidiabetika nur als Teil einer umfassenderen Strategie sehen, bei der die Kontrolle aller vaskulären Risikofaktoren wesentlich für den Erhalt der kognitiven Fähigkeiten ist.
Resümee
Die physiologische Bedeutung von Insulin für unser Gehirn wird immer deutlicher. Insulin ist an der Glukose-Utilisation, der synaptischen Plastizität, der Neuroinflammation, der Beta-Amyloid- Toxizität und an der Tau-Pathologie beteiligt und somit wesentlicher Baustein in der Alzheimer-Pathologie. Trotzdem haben bisher alle therapeutischen Strategien mit intranasalen Insulinen und Insulinsensitizern wie Rosiglitazon und Pioglitazon in randomisierten multizentrischen DB-Studien bei Alzheimer-Krankheit versagt.

AutorIn: Priv.-Doz. Dr. Michael Rainer
Zentrum für geistige Gesundheit und Karl- Landsteiner-Institut für Gedächtnis- und Alzheimerforschung, Wien

Ursprünglich erschienen:
DF 05|2020
DF 05|2020
Herausgeber: Österreichische Diabetes Gesellschaft, Univ.-Prof. Dr. Guntram Schernthaner
Publikationsdatum: 2020-11-27
Zur Ausgabe »
Publikationsdatum: 2020-11-27
Zur Ausgabe »