





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Diabetes und Demenz – Wie vorbeugen, wie behandeln?
5. Oktober 2012

Als in den 1920er-Jahren Insulin zur Behandlung des Diabetes mellitus eingesetzt werden konnte, schien die Heilung dieser Krankheit möglich. Bald stellte sich heraus, dass trotz dieser Therapie Folgeschäden an Nieren, Augen, Gefäßsystem, Herz und peripherem Nervensystem auftreten, die zu Behinderung und Tod führen. Nachdem durch zunehmend wirksame Behandlungsstrategien in den letzten Jahrzehnten diesbezüglich deutliche Fortschritte erzielt wurden, zeigt sich nun, dass die Erkrankung auch Auswirkungen auf andere Organsysteme hat: Knochen und Gehirn. Die zerebralen Folgeschäden rücken dabei zunehmend in den Fokus der Forschung, da Defizite in der kognitiven Leistungsfähigkeit die Lebensqualität der Betroffenen und ihres Umfeldes nachhaltig beeinträchtigen.
Epidemiologische Aspekte
Die zunehmende Lebenserwartung führt zu einer deutlichen Zunahme demenzieller Syndrome. Aktuelle Schätzungen beziffern die Zahl Demenzkranker weltweit mit 24 Millionen, wobei sich diese Zahl verdoppeln wird, wenn die Babyboom-Generation das Alter mit dem größten Risiko für Demenz und kognitiven Abbau erreicht. Auch die Zahl der Typ-2-Diabetiker ist als Folge von Fehlernährung und Bewegungsmangel stark im Steigen begriffen – in den Vereinigten Staaten sind bereits 20 % der über 60-Jährigen betroffen.
Im Jahr 2000 litten in Österreich etwa 100.000 Personen an einem demenziellen Syndrom, Hochrechnungen lassen für 2050 ca. 260.000 Demenzkranke erwarten. Dabei stellt die Alzheimer-Erkrankung (DAT) die häufigste Demenzform dar (60–70 %), gefolgt von der vaskulären Demenz (vaD, 15–25 %) und der Lewy-Body-Demenz (LBD, 6–20 %). Andere Demenzformen sind selten und machen einen Anteil von maximal 10 % aus, während Mischformen besonders im höheren Lebensalter häufig sind.
Die Alzheimer-Erkrankung verläuft chronisch progredient und beginnt meist mit einer Störung des episodischen Gedächtnisses. Im Verlauf treten Defizite in anderen kognitiven Domänen hinzu und beeinträchtigen Sprache, Rechnen, Planen, Problemlösen und räumlich-konstruktive Fähigkeiten. Pathologisch-anatomische Kernmerkmale sind die extrazellulären Amyloid-Plaques (Alzheimer-Plaques) sowie intrazellulär gelegene Ablagerungen paariger Filamente als Folge einer Hyperphosphorylierung des Tau-Proteins (neurofibrilläre Bündel oder „tangles“, NFT). Daneben finden sich Zeichen einer Amyloid-Angiopathie, chronisch-oxidativer und inflammatorischer Schäden sowie mitochondrialer und synaptischer Dysfunktion (Querfurth et al., N Engl J Med 2010).
Die vaskuläre Demenz ist demgegenüber ätiologisch nicht einheitlich definiert, man unterscheidet die Multiinfarktdemenz, die Demenz bei subkortikaler atherosklerotischer Leukenzephalopathie sowie Demenzen nach intrazerebraler Blutung oder im Rahmen von Vaskulitiden.
Störungen kognitiver Funktionen, die keine relevanten Auswirkungen auf die Alltagsfähigkeiten der Betroffenen haben, aber in kognitiven Tests nachweisbar sind, werden unter der Diagnose milde kognitive Beeinträchtigung („mild cognitive impairment“, MCI) zusammengefasst.
Aufgrund der raschen Zunahme von Inzidenz und Prävalenz stellen demenzielle Syndrome eine enorme Herausforderung für das Gesundheitswesen dar, der teilweise durch Entwicklung präventiver Strategien begegnet werden kann: Ein Hinauszögern des Krankheitsbeginns um 5 Jahre könnte die Prävalenz um 50 % reduzieren.
Spezifische Demenzrisikofaktoren bei Diabetes
In Beobachtungsstudien konnten vaskuläre Risikofaktoren wie Hypertonie, Diabetes, Hypercholesterinämie, metabolisches Syndrom und Adipositas in unterschiedlichem Ausmaß mit einem erhöhten Demenzrisiko assoziiert werden. Für die meisten vaskulären Risikofaktoren wurde eine nichtlineare Beziehung zum Auftreten von kognitivem Abbau und Demenz gezeigt, wobei für das mittlere Lebensalter die stärksten Assoziationen bestehen (Kloppenborg et al., Eur J Pharmacol 2008). Dies betont die wesentliche Rolle der Expositionsdauer gegenüber einem Risikofaktor, aber auch, dass diese Faktoren tatsächlich als unabhängige Risikofaktoren zu werten sind und keine Marker einer möglicherweise zugrunde liegenden gemeinsamen Ätiopathogenese. Auch Depression und Schädel-Hirn-Trauma sind mit einem erhöhten Risiko verbunden, wohingegen körperlichen, geistigen und sozialen Aktivitäten präventive Effekte attribuiert werden.
Die konsequente Kontrolle vaskulärer Risikofaktoren und das Aufrechterhalten eines aktiven Lebensstils scheinen nach derzeitigem Kenntnisstand das Auftreten und Fortschreiten von kognitivem Abbau und Demenz zu verzögern, allerdings ist die Datenlage noch inkonsistent. Um den Einfluss der Modifikation von Risikofaktoren auf die Endpunkte kognitiver Leistungsfähigkeit und Demenz zu erfassen, sind prospektive Interventionsstudien zu fordern.
Abbildung 1 fasst die möglichen Einflussfaktoren zusammen:
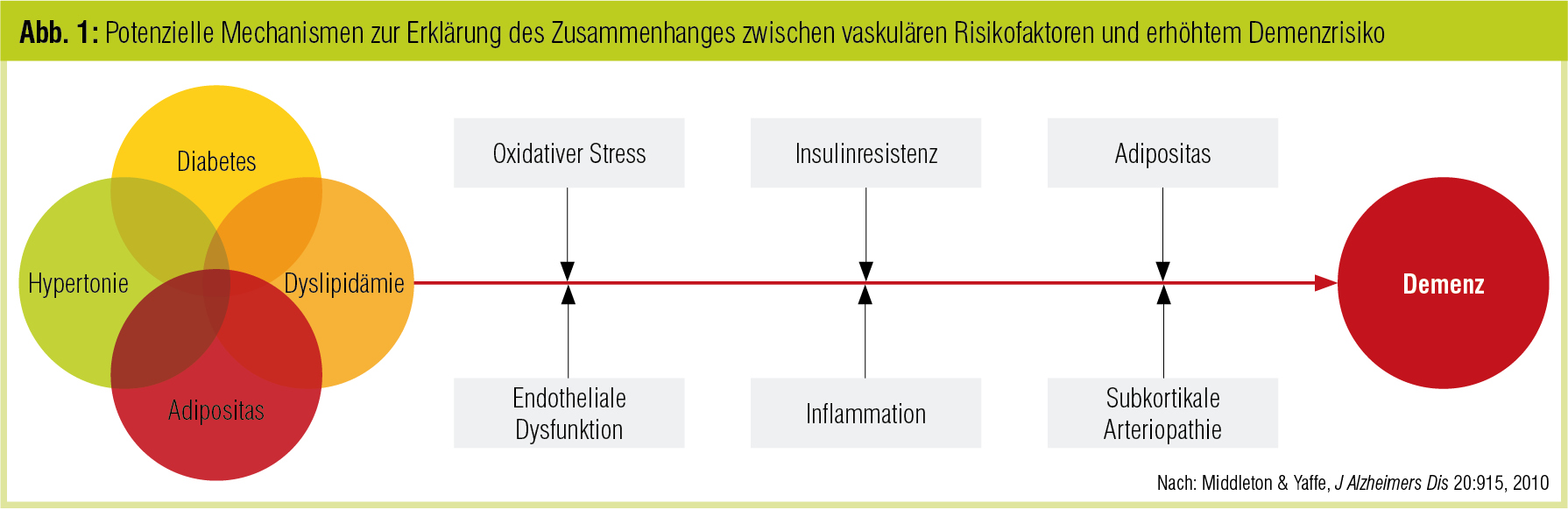
Nach longitudinalen Studien haben Patienten mit Typ-2-Diabetes über einen Beobachtungszeitraum von 2–8 Jahren ein 1,5- bis 2-fach höheres Risiko für eine kontinuierliche Abnahme kognitiver Funktionen, das Ausmaß der jährlichen Verschlechterung kognitiver Leistungen ist im Vergleich zu Stoffwechselgesunden um 20–50 % höher (Biessels et al., Lancet Neurol 2006). Dauer sowie Schwere des Typ-2-Diabetes korrelieren mit Ausmaß und Progredienz des kognitiven Abbaus (Yaffe et al., Arch Neurol 2012). Eine aktuelle Studie zeigt allerdings, dass funktionell unabhängige Menschen mit Typ-2-Diabetes offensichtlich nur einen sehr geringen kognitiven Abbau erleiden: In einem 4-jährigen Beobachtungszeitraum unterschieden sich die kognitiven Einbußen nicht statistisch signifikant von Nichtdiabetikern (van den Berg et al., Diabetologia 2010). Dies kann dahin gehend interpretiert werden, dass eine lange Exposition gegenüber dem Risikofaktor Diabetes erforderlich ist, um Auswirkungen auf kognitive Funktionen zu generieren, was nicht verwundert, da bei degenerativen Demenzen neuropathologische Veränderungen bereits 20–30 Jahre vor klinischer Manifestation nachweisbar sind und somit eine lange subklinische Phase postuliert wird.
Auch in einem hochaltrigen Kollektiv erhöhte ein Typ-2-Diabetes das Risiko, an einer Demenz – insbesondere an vaskulärer Demenz – zu erkranken. Dieses Risiko wurde durch eine schwere Hypertonie oder Herzerkrankung noch deutlich akzentuiert. Aktuelle Publikationen sorgen aber auch hier für eine inkonsistente Datenlage. So konnte an einem Kollektiv mit Alzheimer-Demenz gezeigt werden, dass das Vorhandensein eines Diabetes mellitus mit einem verzögerten kognitiven Abbau assoziiert war. Als mögliche Erklärung geben die Autoren die potenziell konsequentere Behandlung von vaskulären Risikofaktoren bei Diabetikern an (Sanz et al., Neurology 2009).
Auch milde kognitive Beeinträchtigungen sind bei Menschen mit Diabetes häufiger. Ebenso sind gestörte Glukosetoleranz sowie Prädiabetes mit einem erhöhten Risiko für das Auftreten einer Demenz jedweder Ätiologie vergesellschaftet.
Pathophysiologische Zusammenhänge
Insulinresistenz und Insulin-Signalling. Rezente Publikationen legen nahe, dass die Hyperinsulinämie sowohl die Amyloid-Beta-Deposition (Aβ-Deposition) als auch die Phosphorylierung des Tau-Proteins beeinflusst – also beide derzeit als zentral geltenden Mechanismen der Pathogenese der Alzheimer-Demenz (Biessels et al., Lancet Neurol 2006). Eine künstlich erzeugte moderate Hyperinsulinämie kann Entzündungsmarker (F2-Isoprostane und TNF-alpha;) sowie Aβ-Konzentrationen – und zwar sowohl im peripheren Blut als auch im Liquor cerebrospinalis – erhöhen. Diese Beobachtungen können als Hinweise auf eine wesentliche Rolle entzündlicher Mechanismen für das erhöhte Demenzrisiko bei Diabetes interpretiert werden.
Die Insulinresistenz führt zu einer verminderten Synthese des „insulin-degrading enzyme“ (IDE), das auch für den Abbau von Aβ verantwortlich ist. Eine verminderte Enzymaktivität kann daher zu einer vermehrten Aβ-Ablagerung führen: In einer neuropathologischen Studie konnte nachgewiesen werden, dass Insulinresistenz und Hyperinsulinämie mit einer Zunahme der Zahl der Alzheimer-Plaques einhergehen, besonders bei Trägern des APOEε4-Allels, welches per se ein höheres Alzheimerrisiko definiert. In einer weiblichen Population war das Ausmaß der Insulinresistenz, definiert über den HOMA-Index, negativ mit dem Volumen des Hippokampus korreliert, der als zentrales anatomisches Korrelat des episodischen Gedächtnisses angesehen wird. Manche Autoren räumen der zerebralen Insulinresistenz und ihrem Bezug zur Alzheimer-Demenz einen hohen ätiopathogenetischen Stellenwert ein und sprechen von einem „brain diabetes“ oder Diabetes mellitus Typ 3.
Mikroangiopathie. Ein gestörtes Insulin-Signalling induziert möglicherweise eine Zunahme der Aktivität der Glykogen-Synthase-Kinase-3β mit einer gesteigerten Phosphorylierung des Tau-Proteins und vermehrter NFT-Bildung. Demgegenüber finden sich bei dementen Diabetikern in autoptischen Serien weniger histopathologische Merkmale der Alzheimer-Demenz als bei Individuen ohne Diabetes. Es liegt daher nahe, dass asymptomatische ischämische Läsionen als Folge der „small vessel disease“ bei älteren Diabetikern die Schwelle für das Auftreten einer Demenz erniedrigen können. Dies könnte die Inkonsistenz zwischen den Ergebnissen der Grundlagenforschung und neuropathologischen Untersuchungen erklären. Eine erhöhte Prävalenz von stummen Infarkten und „white matter lesions“ (WML) konnte bei Typ-2-Diabetes wiederholt gezeigt werden. Bei identischem Level des kognitiven Abbaus finden sich bei den Individuen, die mit zwei ätiopathogenetischen Faktoren belastet sind, diese in jeweils geringerer Ausprägung als beim Vorliegen nur einer Ätiologie.
Hypoglykämien. Wiederholte und protrahierte Hypoglykämien können ebenfalls zur Pathogenese des kognitiven Abbaus bei Diabetes beitragen, wie in Studien bei Patienten mit Typ-1-Diabetes gezeigt werden konnte. Auch bei Typ-2-Diabetes scheinen schwere Hypoglykämien das Demenzrisiko zu erhöhen, während die Rolle leichter hypoglykämischer Episoden nicht vollständig geklärt ist. Länger anhaltende Hypoglykämien können jedenfalls zu irreversiblen Gehirnschäden beitragen. An jüngeren Typ-1-Diabetikern konnte neben einer leichten zerebralen Atrophie gezeigt werden, dass in Hirnregionen, die für Sprachprozesse und Gedächtnis relevant sind, eine etwas geringere Dichte der grauen Substanz vorliegt. Abbildung 2 fasst diese Veränderungen sowie relevante Risikofaktoren zusammen.
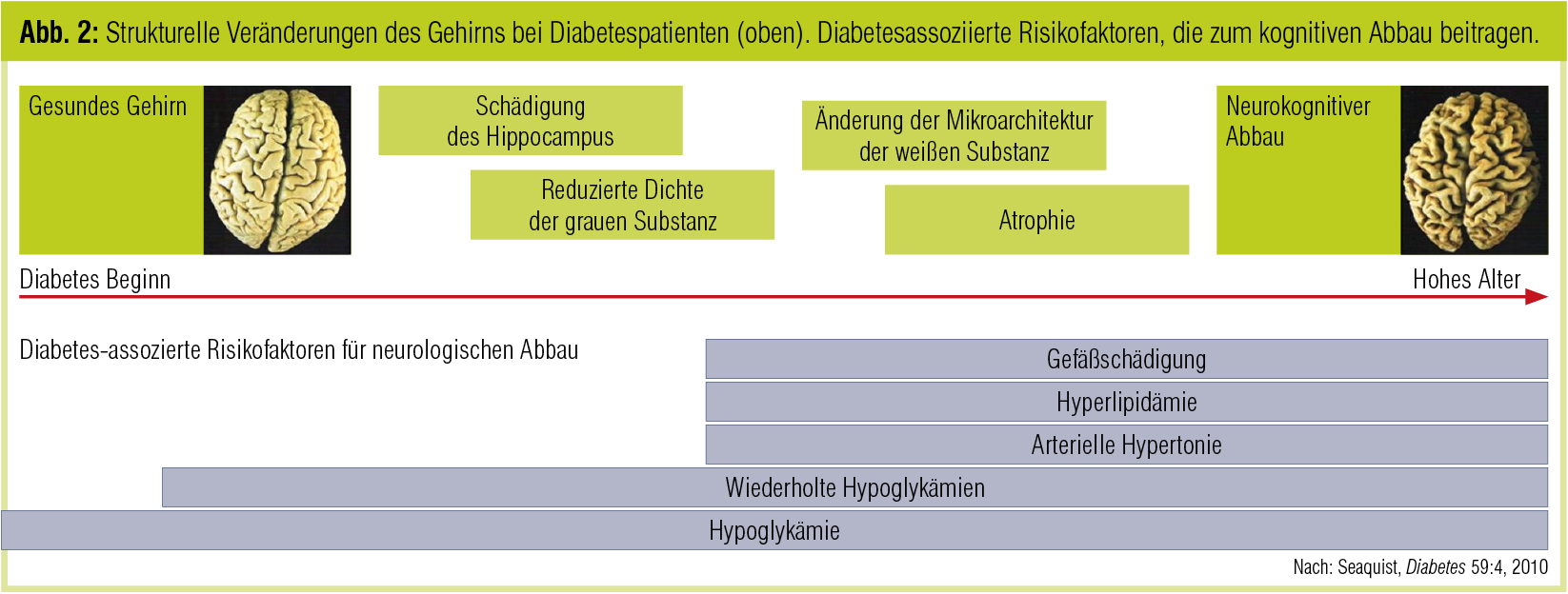
Oxidativer Stress. Zunehmende Aufmerksamkeit erlangen die im Kohlehydratstoffwechsel anfallenden „advanced glycation end-products“ (AGE). Histopathologische Untersuchungen von Alzheimer-Kranken mit Typ-2-Diabetes zeigten neben der Alzheimer-Pathologie auch erhöhte AGE-Spiegel, eine Zunahme von RAGE-positiven („receptor for AGE“) Zellen und verstärkte Aktivierung von Mikroglia. Nach der Bindung löst der RAGE im Zellinneren eine Kette von Reaktionen aus und aktiviert auch den Transkriptionsfaktor NF-κB. Dieser vermittelt die Bildung von Proteinen, die Entzündungsprozesse und Abwehrreaktionen im Körper auslösen. RAGE erkennt auch andere Peptide, die bei chronisch-entzündlichen und altersbedingten Erkrankungen vermehrt gebildet werden – dazu zählt auch Aβ. Somit kann eine Zunahme der Zellschädigung durch einen AGE-getriggerten Circulus vitiosus oxidativer Stressmechanismen postuliert werden (Guglielmotto et al., Neurobiol Aging 2012).
Weitere Mechanismen der Beeinträchtigung der Gedächtnisleistung durch Insulinresistenz, Hyperinsulinämie und Typ-2-Diabetes werden diskutiert (Watson & Craft, CNS Drugs 2003): Dazu zählen verminderte zentralnervöse Insulinspiegel, eine verstärkte Entzündungsantwort (IL-6, TNF-alpha;), Veränderungen des zerebralen Glukosemetabolismus, Veränderungen der Neurotransmitterexpression bzw. aktivität und letztlich auch Veränderungen der Langzeitpotenzierung (LTP), die als Korrelat des synaptischen Lernens angesehen wird.
Das Inkretin Glucagon-like peptide-1 (GLP-1) unterstützt das Insulin-Signalling bei Typ-2-Diabetes. Im Mausmodell konnte gezeigt werden, dass GLP-1-Rezeptor-Knock-out-Tiere in der räumlichen Erinnerung eine deutlich schlechtere Lernleistung zeigten, was durch eine massive Beeinträchtigung der LTP im Hippokampus erklärt werden konnte. Zumindest im Tiermodell kann daraus ein potenziell neuroprotektiver Effekt von GLP-1-Analoga abgeleitet werden.
Möglichkeiten der Demenzprävention
Ob eine optimale glykämische Kontrolle auch zur Verringerung des kognitiven Abbaus oder der Demenzinzidenz beiträgt, kann aufgrund des Fehlens prospektiver Studien mit entsprechenden Endpunkten nicht definitiv gesagt werden, über den Weg der Prävention zerebrovaskulärer Ereignisse kann jedoch grundsätzlich davon ausgegangen werden. Auch konsequente Diät und vermehrte körperliche Aktivität lassen einen positiven Effekt erwarten. So konnte im Tiermodell gezeigt werden, dass körperliche Aktivität sowohl zu einer Anregung der Neurogenese im Hippokampus als auch zu einer verstärkten dendritischen Verschaltung führt. Körperliche und geistige Aktivität sind darüber hinaus mit einer verminderten Bildung von Amyloid-Plaques assoziiert.
Angesichts der Bedeutung der zerebralen Insulinresistenz ist eine antidiabetische Therapie eine mögliche Strategie für die Prävention und Behandlung von Demenzen. Der potenziell neuroprotektive Effekt von GLP-1-Analoga wurde bereits erwähnt. Studien mit Agonisten des Peroxisome proliferator-activated receptor γ (PPAR-γ) konnten positive Effekte auf Merkfähigkeit und selektive Aufmerksamkeit zeigen. Für Metformin und Sulfonylharnstoffe liegen Studiendaten vor, die zeigen, dass die kombinierte Therapie mit diesen Substanzen das Risiko, in einem Zeitraum von 8 Jahren an Alzheimer-Demenz zu erkranken, reduzieren kann (Hsu et al., J Alzheimers Dis 2011). Auch intranasal appliziertes Insulin – das vorrangig zentralnervöse Wirkung entfaltet – führte in Studien zu einer Besserung der verbalen Merkfähigkeit (Morris et al., Curr Neurol Neurosci Rep 2012).

Ursprünglich erschienen:
DF 04|2012
DF 04|2012
Herausgeber: Univ.-Prof. Dr. Guntram Schernthaner, Österreichische Diabetes Gesellschaft
Publikationsdatum: 2012-10-05
Zur Ausgabe »
Publikationsdatum: 2012-10-05
Zur Ausgabe »