





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Statine und Diabetesrisiko – Bei höherem Risiko überwiegt der Nutzen
28. August 2012
Die klinische Entwicklung der HMG-CoA-Reduktase-Inhibitoren (Statine) stellt wahrscheinlich den größten Fortschritt in der kardiovaskulären Pharmakologie der vergangenen Jahrzehnte dar. Die Metaanalyse der Cholesterol Treatment Trialists’ (CTT) Collaboration (Lancet 2005) an ca. 90.000 Patienten aus 14 randomisierten Studien zeigt, dass eine Senkung des LDL-Cholesterins um ca. 40 mg/dl zu einer relativen Absenkung der Myokardinfarkte um 23 %, der koronaren Revaskularisationen um 24 %, der Schlaganfälle um 17 % und der Gesamtsterblichkeit um 12 % führt. Damit gehören die Statine zu der auserlesenen Handvoll von Medikamenten mit einem günstigen Effekt auf die Sterblichkeit bei „Volkskrankheiten“ wie manifester Atherosklerose, Diabetes mellitus oder anders bedingtem hohem kardiovaskulärem Risiko.
Gerade als wir alle uns an die angenehme Idee, dass der Nutzen der Statine lediglich mit geringfügigen und/oder seltenen Nebenwirkungen erkauft werden muss, gewöhnt hatten, ergibt sich mit dem Nachweis der durch Statine bedingten erhöhten Diabetesinzidenz doch eine gewisse Änderung der Sachlage.
Evidenz für vermehrte Diabetesfälle unter Statintherapie
Diabetesinzidenz bei Verwendung von Statinen vorwiegend in Standardintensität bei klinisch manifester Atherosklerose oder hohem kardiovaskulärem Risiko. Eine von Sattar et al. (Lancet 2010) erstellte Metaanalyse umfasst 13 Statinstudien mit insgesamt 91.140 zu Beginn nichtdiabetischen Patienten, von denen 4.278 über einen mittleren Beobachtungszeitraum von 4 Jahren einen Diabetes entwickelten. Lediglich Studien mit mehr als 1.000 Teilnehmern und einer Studiendauer von mehr als einem Jahr wurden inkludiert; bereits zu Studienbeginn diabetische Patienten wurden für weitere Berechnungen nicht berücksichtigt. Nachdem die Studien für den Endpunkt Diabetesinzidenz nicht ausgelegt waren, ist die Diabetesdiagnose höchst variabel („adverse event report“ von Diabetes, Beginn einer antidiabetischen Medikation oder Nüchternblutzucker > 126 mg/dl).
Obwohl nur 2 der 13 individuellen Studien einen signifikanten Anstieg der Diabetesneuerkrankungen zeigten, fand sich in der Metaanalyse ein signifikanter Anstieg der Diabetesrate um 9 % (95%-KI: 2–17 %; Tab. 1). Der Unterschied fand sich unabhängig davon, ob Patienten im Kontrollarm Placebo oder „usual care“ erhielten, und er war – interessanterweise – nicht unterschiedlich zwischen lipophilen (Odds Ratio [OR] = 1,10) und hydrophilen (OR = 1,08) Statinen. Der Versuch, Risikofaktoren für die Diabetesentstehung zu identifizieren, ergab einen signifikant höheren Risikogradienten zwischen Statin und Placebo/„usual care“ bei höherem Alter sowie einen Trend zu einem höheren Gradienten bei höherem Body Mass Index (BMI) und höherer Absenkung des LDL-Cholesterins (LDL-C).
Das absolute Risiko jedoch war niedrig – es mussten 255 Patienten für 4 Jahre behandelt werden, um einen zusätzlichen Diabetesfall zu verursachen. Die „number needed to harm“ (NNH), bezogen auf 1 Jahr Behandlungsdauer, betrug also 1.020. Die „number needed to treat“ (NNT) derselben Population zur Verhinderung eines kardiovaskulären Ereignisses bezogen auf 1 Jahr Behandlungsdauer liegt bei Patienten mit manifester koronarer Herzkrankheit (KHK) bei 104, bei Patienten ohne manifester KHK bei 200. Wichtig für das Verhältnis von NNT zu NNH sind die Cholesterinausgangswerte: Nachdem eine Standardstatintherapie eine ca. 30%ige LDL-C-Absenkung bewirkt, entsteht bei höherem Cholesterinausgangswert eine stärkere Reduktion vaskulärer Ereignisse bei gleichem diabetogenem Potenzial, d. h. höherer Nutzen bei gleichem Risiko.

Zusammenfassend ergibt sich für Standardstatine bei manifester KHK für jeden „zusätzlichen“ Diabetesfall zumindest eine Verhinderung von 10 kardiovaskulären Ereignissen, davon 5 Myokardinfarkte. Standardstatine bei hohem kardiovaskulärem Risiko ohne manifeste KHK verhindern für jeden „zusätzlichen“ Diabetesfall zumindest 5 kardiovaskuläre Ereignisse.
Diabetesinzidenz bei Verwendung von Statinen „hoher Potenz“ bei manifester Atherosklerose. Zwischenzeitlich ergab sich 2011 die Nachanalyse der SPARCL-Studie (Waters et al., J Am Coll Cardiol 2011), in der Patienten in der chronischen Phase nach einem ischämischen Schlaganfall oder einer transitorischen ischämischen Attacke (TIA) auf Atorvastatin 80 mg vs. Placebo randomisiert und 4,9 Jahre beobachtet wurden. Von 3.803 Patienten ohne Diabetes entwickelten 166 auf Atorvastatin und 115 auf Placebo einen Diabetes – ein Anstieg um 44 % (NNH 75).
Zusammenfassend ergibt sich für Atorvastatin 80 mg bei „Post-stroke“-Patienten für jeden „zusätzlichen“ Diabetesfall eine Verhinderung von ca. einem (1,4) kardiovaskulären Ereignis.
Die naheliegende Frage, ob diese höhere Diabetesrate Ausdruck der potenteren Statintherapie war, konnten Preiss et al. (JAMA 2011) anhand einer Metaanalyse von 5 Statinstudien mit 32.752 initial nichtdiabetischen Patienten, in denen hochpotente mit weniger potenten Statinregimes (z. B. in der TNT-Studie Atorvastatin 80 mg vs. Atorvastatin 10 mg) verglichen wurden, beantworten. Wiederum fand sich im intensiveren Therapiearm ein signifikanter Anstieg der Diabetesrate um 12 % (95%KI: 4–22 %; Tab. 2). Die NNH, bezogen auf 1 Jahr Behandlungsdauer für den Vergleich hochpotentes Statin vs. Standardstatin, betrug in diesem Fall 498. In diesem Vergleich stellten höheres Alter und höherer BMI keine Indikatoren für einen höheren Riskogradienten dar. Ebenso fand sich kein Unterschied im Diabetesrisiko zwischen den beiden intensiven Prüfregimes (Atorvastatin 80 mg: OR = 1,15; Simvastatin 80 mg: OR 1,13) oder zwischen Studien beim ACS (OR 1,15) oder bei stabiler KHK (OR = 1,12).
Von praktischem Wert dürfte der Vergleich von Nutzen und Risiko einer intensiven Statintherapie vs. einer Standardstatintherapie sein. Die jährliche NNT beträgt in dieser Metaanalyse mit ausschließlich KHK-kranken Patienten zur Verhinderung einer koronaren Revaskularisation 171, zur Verhinderung eines nichttödlichen Myokardinfarkts 578, zur Verhinderung eines nichttödlichen Schlaganfalls 1.538 und zur Verhinderung eines kardiovaskulären Todesfalls 578. Im Vergleich dazu beträgt in dieser Kohorte die jährliche NNH, welche zur Entstehung eines zusätzlichen Diabetesfalls führt, 498.
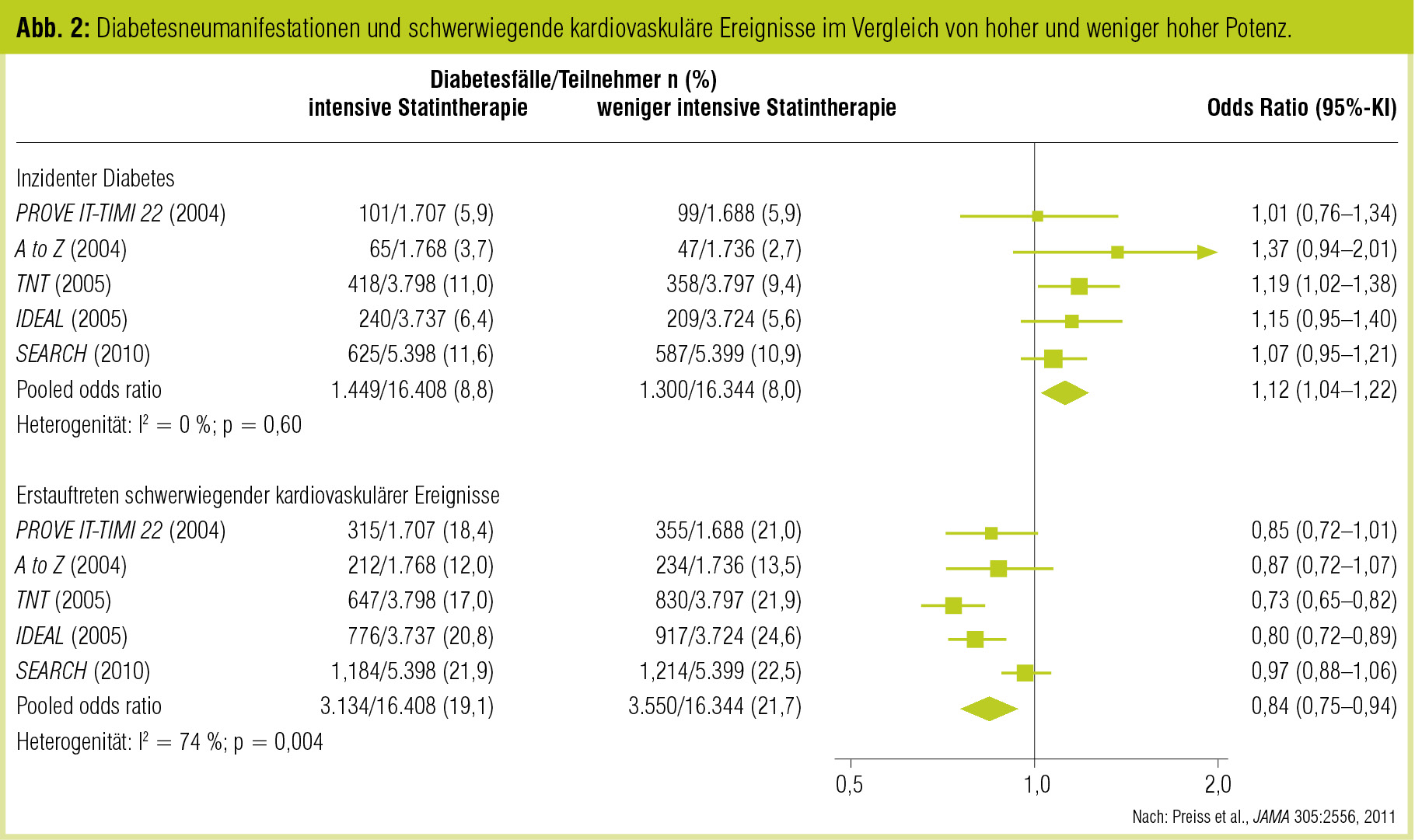
Zusammenfassend ergibt sich für hochpotente Statine vs. Standardstatine bei KHK für jeden „zusätzlichen“ Diabetesfall eine Verhinderung von zumindest 3 kardiovaskulären Ereignissen.
Diabetesinzidenz bei Verwendung von Statinen „hoher Potenz“ ohne manifeste Atherosklerose („Primärprävention“) In der JUPITER-Studie (Ridker et al., New Engl J Med 2008) wurden 17.802 als „klinisch gesund“ beschriebene Männer (> 50 Jahre) und Frauen (> 60 Jahre) mit einem LDL-C 2 mg/l) für ca. 2 Jahre mit Rosuvastatin 20 mg oder Placebo behandelt. Für die Wertung dieser Studie wichtig ist, dass ca. 40 % der Teilnehmer die NCEP-Kriterien des metabolischen Syndroms erfüllten, sich also in einem prädiabetischen Zustand befanden. Diabetes war ein Ausschlusskriterium. Das absolute kardiovaskuläre Risiko in dieser Studie war mäßig bis gering (8,5 % Myokardinfarkt, Schlaganfall und kardiovaskulärer Tod pro 10 Jahre. Zum Vergleich: Als hohes kardiovaskuläres Risiko wird gemäß Framingham-Score eine > 20%-Wahrscheinlichkeit für Myokardinfarkt oder plötzlichen Herztod über 10 Jahre definiert). In der Tat bewirkte die potente Statintherapie (LDL-C-Senkung ca. 50 %) selbst in dieser Niedrigrisikokohorte eine Halbierung der kardiovaskulären Ereignisse und eine signifikante Reduktion der Gesamtsterblichkeit um 20 %. Neu diagnostizierter Diabetes trat bei 3 % der mit Statin behandelten Patienten vs. 2,4 % der mit Placebo behandelten Patienten auf (+25 %; p = 0,01). Einer NNT/Jahr von 169 zur Verhinderung eines (breit gefassten) primären vaskulären Endpunktes steht also in JUPITER eine NNH/Jahr von 167 im Sinn von statininduziertem Diabetes gegenüber.
Zusammenfassend ergibt sich für Rosuvastatin 20 mg bei Patienten mit mäßigem bis geringem kardiovaskulärem Risiko sowie einer hohen Rate von „Prädiabetes“ für jeden „zusätzlichen“ Diabetesfall eine Verhinderung von ca. einem kardiovaskulären Ereignis.
Beeinflussung der Glykämie durch Statine
Patienten mit Typ-2-Diabetes. Statine weisen dramatische Wirksamkeit in der Prävention vaskulärer Ereignisse bei Diabetespatienten sowohl mit als auch ohne bereits manifester Atherosklerose auf (z. B. HPS, CARDS, diabetische Subgruppe in TNT). Nachdem bei Typ-2-Diabetes eine zunehmende Verschlechterung der Betazellrestfunktion eintreten kann und die Behandler vor zunehmende therapeutische Probleme in der Diabeteskontrolle stellt, ist die Frage naheliegend, ob Statine diesen Prozess unter Umständen ungünstig beeinflussen. Trotz enormer und langjähriger Studienerfahrung bestehen derzeit keine klaren Daten, die diese potenzielle Sorge untermauern könnten. So konnten z. B. in HPS und CARDS keine HbA1c-Unterschiede zwischen Statin- und Kontrollgruppe festgestellt werden.
Ohne eine exakte Aufarbeitung etwaiger Änderungen in der Intensität der medikamentösen und/oder diätetischen Diabetestherapie kann diese Frage allerdings nicht abschließend beantwortet werden.
Patienten ohne Typ-2-Diabetes. Die Analyse der HbA1c-Werte von nichtdiabetischen Personen zeigt lediglich einen minimalen, wenn auch ungünstigen Effekt von Statinen. In JUPITER fand sich in der Rosuvastatin-Gruppe nach 2 Jahren ein HbA1c von 5,9 % vs. 5,8 % auf Placebo (p = 0,001). Auch hier findet sich, analog zur Diabetesinzidenz, eine Dosis-Wirkung-Beziehung innerhalb der Statine. In PROVE IT-TIMI 22 (Sabatine et al., Circulation 2004) fand sich in der Gruppe mit intensiver Statintherapie (Atorvastatin 80 mg) ein um 0,19 Prozentpunkte höheres HbA1c als in der Standardgruppe (Pravastatin 40 mg). Ebenso dokumentieren viele Statinstudien ein geringfügig höheres Körpergewicht in der Statingruppe (+0,5–1 kg).
Mechanistische Daten zur Beeinflussung der Glykämie beim Menschen durch Statine sind widersprüchlich. Zwei Untersuchungen unter Verwendung der hyperinsulinämischen, euglykämischen Klemmtechnik, des Goldstandards in der Evaluation der Insulinresistenz, zeigten keine Beeinflussung durch Statine. Koh et al. (J Am Coll Cardiol 2010) hingegen untersuchten 213 hypercholesterinämische koreanische Patienten vor und 2 Monate nach Behandlung mit Atorvastatin (10, 20, 40 und 80 mg) im Vergleich zu einer Placebo-Kontrollgruppe. Es fanden sich ein signifikanter HbA1c-Anstieg von 0,2–0,3 Prozentpunkten sowie eindrucksvolle Steigerungen der Insulinkonzentration von 25 bis 45 % durch Atorvastatin. Die Tatsache, dass es sich hierbei um eine Single-Center-Studie an asiatischen Patienten handelt, die fehlende Dosis-Wirkung-Beziehung, das gerade bei dieser Fragestellung eigenartig bunt gemischte Studienkollektiv (glukosenormal, glukoseintolerant bis diabetisch) sowie die grenzwertigen absoluten Effekte machen eine Reproduktion dieser Daten obligat.
Zusammenfassend führen Statine bei Patienten ohne Typ-2-Diabetes zu einer geringgradigen HbA1c-Steigerung um 0,1–0,3 Prozentpunkte, wobei potente Statine hier ungünstiger als Standardstatine zu sein scheinen. Laut UKPDS würde eine HbA1c-Steigerung von 0,2 Prozentpunkten bei frühem Typ-2-Diabetes einen Anstieg der mikroangiopathischen Diabeteskomplikationen von 5 % auslösen.
Wie bewirken Statine einen Anstieg der Diabetesinzidenz?
Die für klinisch tätige Ärzte günstigsten Formen der Kausalität, nämlich Überlebensvorteil („survival bias“, d. h. statinbehandelte Patienten leben länger und haben deshalb eine höhere Chance, Diabetes zu entwickeln) und „ascertainment bias“ (d. h. statinbehandelte Patienten haben mehr – triviale – unerwünschte Wirkungen und im Rahmen der dadurch bedingten Arztkontakte eine höhere Chance einer Diabetesdiagnose) treffen nicht zu. „Lifestyle bias“ (d. h. Lockerung der Diät durch günstige Lipidmesswerte) ist bei gegebener Verblindung von Cholesterinwerten (zumindest die Studienärzte und Patienten betreffend) in randomisierten Studien unplausibel.
Eine biochemische Basis der diabetogenen Wirkung von Statinen ist also naheliegend. Prinzipiell weisen Statine sowohl potenziell antidiabetogene als auch diabetogene molekulare Effekte auf: Antidiabetogene Wirkungen wären z. B. Antiinflammation, Erhöhung von Stickstoffmonoxid, Reduktion der Lipotoxizität durch Triglyzeridabsenkung und Erhöhung des HDL-Cholesterins sowie die günstige Beeinflussung von Adipokinen (z. B. Adiponektin). Diabetogene Wirkungen könnten wiederum verschiedene Organ betreffen: die Leber als das für die Lipidsenkung der Statine wichtigste Organ, den Muskel als Organ relevanter Statinnebenwirkungen sowie das Fettgewebe. In der Tat zeigen verschiedene experimentelle Systeme, z. T. unter Verwendung exzessiver Statindosen, die Induktion zellulärer Insulinresistenz in Leber, Muskel und/oder Fettgewebe.
Derzeit am schlüssigsten kann die Steigerung der Diabetogenese allerdings durch einen unerwünschten Statineffekt an der Betazelle erklärt werden: Bekanntermaßen ist Cholesterin für das Funktionieren aller menschlichen Zellen insofern unentbehrlich, als es durch Einbau in die Phospholipid-Bilayer der Plasmamembran und anderer subzellulärer Membranen die Fluidität und damit die Funktionalität vieler Proteine (z. B. Transport-, Rezeptor- und Signaltransduktionsproteine) steuert. Paradigmatisch für diese Situation sind besonders cholesterinreiche Mikrodomänen der Plasmamembran, so genannte „lipid rafts“. Die Betazelle ist nunmehr dadurch gekennzeichnet, dass sie über ca. 9.000 Insulin enthaltende sekretorische Granula verfügt, welche insgesamt ca. das Fünffache der Oberfläche der Plasmamembran ausmachen. Diese Situation macht die Betazelle besonders empfindlich für Störungen der endogenen Cholesterinbiosynthese, wie anhand von genetischen Störungen (z. B. Smith-Lemli-Opitz-Syndrom oder selbst heterozygoter ABCA1-Defizienz/Tangier-Erkrankung) beim Menschen oder im Tierversuch (Gene-targeting-Experimente an ABCG1(-/-)-Mäusen) gezeigt wurde.
Zusammenfassend führen sowohl eine Reduktion des Cholesteringehalts der sekretorischen Granula als auch eine Erhöhung des Cholesteringehalts der Plasmamembran der Betazelle zu verminderter Insulinsekretion. Statine könnten Ersteres durch die direkte Hemmung der HMG-CoA-Reduktase in der Betazelle und Letzteres durch indirekte LDL-Rezeptor-Up-Regulation in der Betazelle mit nachfolgender Cholesterinverteilungsstörung zwischen subzellulären Kompartimenten bedingen.
Konsequenzen für die Praxis
Die Erhöhung der Diabetesinzidenz durch Statine ist zwar nicht trivial, wird aber durch 3 Überlegungen relativiert:
- Sehr wahrscheinlich führen Statine nicht monokausal zu Diabetes, sondern enttarnen lediglich genetisch prädisponierte Personen, die ohnehin in Bälde diabetisch geworden wären.
- Mikrovaskuläre Komplikationen stehen in der Frühphase des Typ-2-Diabetes nicht im Vordergrund; stattdessen bestimmen atherosklerotische Komplikationen das Schicksal der Patienten.
- Die ungünstige Auswirkung auf die Prognose von eingetretenen vaskulären Katastrophen (z. B. Myokardinfarkt, Schlaganfall) ist ungleich höher als die einer Neudiagnose von Typ-2-Diabetes.
Eine laufende Statintherapie bei Nichtdiabetikern sollte nunmehr aber Anlass für Glukosekontrollen sein.
Wichtig ist festzuhalten, dass es sich nach derzeitigem Wissen um eine statinspezifische Nebenwirkung und nicht um eine Konsequenz der LDL-Senkung handelt. Daher eröffnen diese Daten neue Chancen für diätetische Zugänge, für Kombinationen von Standardstatinen mit dem Cholesterinresorptionshemmer Ezetimib oder mit Fenofibrat sowie für Pharmainnovationen (z. B. auf apoB, PCSK9 oder CETP gerichtete LDL-senkende Therapien).
Weitere Literatur beim Verfasser

AutorIn: Prim. Univ.-Doz. Dr. Bernhard Föger
Interne Abteilung, Landeskrankenhaus Bregenz

Ursprünglich erschienen:
DF 03|2012
DF 03|2012
Herausgeber: Prim. Univ.-Prof. Dr. Guntram Schernthaner, Österreichische Diabetes Gesellschaft
Publikationsdatum: 2012-06-29
Zur Ausgabe »
Publikationsdatum: 2012-06-29
Zur Ausgabe »