Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
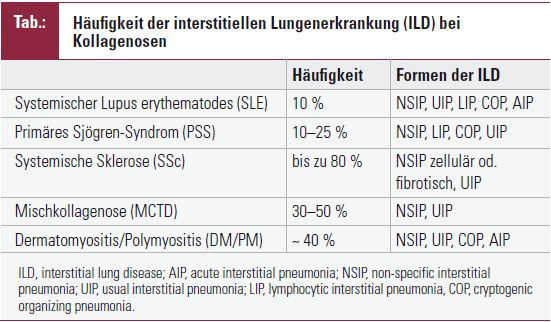
Interstitielle Lungenerkrankung bei Kollagenosen
25. November 2011
Bei den Kollagenosen sind pulmonale Komplikationen häufig. Die interstitielle Lungenerkrankung (ILD) und die pulmonal- arterielle Hypertension (PAH, pulmonale Vaskulopathie) sind die häufigsten und wichtigsten Formen der Lungenbeteiligung. Eine pulmonale Hypertension kann auch als Folge ausgedehnter interstitieller Lungenveränderungen auftreten. Mit Hilfe sensitiver Untersuchungsmethoden können sowohl die ILD als auch die PAH noch vor dem Auftreten von kardiopulmonalen Symptomen erfasst werden. Nachdem eine frühzeitige therapeutische Intervention das Fortschreiten der Lungenerkrankung verhindern kann, sind Untersuchungen im Hinblick auf eine Lungenbeteiligung am Beginn der Erkrankung und im weiteren Verlauf unerlässlich.
Diagnostische Strategien
Neben der genauen Anamnese hinsichtlich kardiopulmonaler Symptome wie Husten, Belastungsdyspnoe, Thoraxschmerz, synkopale Anfälle und einer eingehenden klinischen Untersuchung (Rasselgeräusche, jugulärer Venenpuls, Herztöne, Herzgeräusche) sind die konventionelle Thoraxröntgenuntersuchung, die Lungenfunktionsprüfung einschließlich der Bestimmung der Diffusionskapazität (DLCO), die hochauflösende Computertomografie (HR-CT), die Echokardiografie zur Abschätzung des pulmonal-arteriellen Druckes, das Elektrokardiogramm zur Erfassung einer Rechtsherzbelastung und kardiale Biomarker (NT-proBNP) hilfreich. Bei der Lungenfunktionsprüfung weist eine restriktive Ventilationsstörung in Kombination mit einer Abnahme der DLCO auf eine ILD hin. Eine deutlich herabgesetzte DLCO ohne Veränderung der Lungenvolumina oder ein signifikanter Abfall der DLCO innerhalb eines Jahres können Ausdruck einer PAH sein. Mit Hilfe der HR-CT werden interstitielle Lungenveränderungen erfasst. Milchglasverschattungen (ground-glass opacities) und netzartige, streifige Strukturalterationen (reticular opacities) kennzeichnen eine ILD. Milchglasverschattungen sind meist Ausdruck eines aktiven entzündlichen Prozesses im Sinne einer floriden Alveolitis, können aber auch durch Ödeme, eine beginnende Fibrose oder Infektionen bedingt sein. Dementsprechend kommt bei der Interpretation des radiologischen Befundes dem klinischen Erscheinungsbild und der Verlaufsbeobachtung besondere Bedeutung zu. Zur Sicherung der Diagnose sind unter Umständen weitere, invasive Untersuchungen wie die Bronchiallavage (BAL) und die videoassistierte, thorakoskopische oder offene Lungenbiopsie notwendig. In der Lavage-Flüssigkeit findet man bei der ILD vermehrt neutrophile Granulozyten, aber auch Lymphozyten. Der Nachweis von Granulozyten in der Lavageflüssigkeit wird als Zeichen des aktiven entzündlichen Prozesses aufgefasst. Die Lungenbiopsie kann vor allem zum Ausschluss einer infektiösen Genese (opportunistische Infektionen) notwendig sein.
Histopathologie – prognostische Relevanz
Die kollagenoseassoziierte ILD präsentiert sich klinisch und histopathologisch überwiegend als nichtspezifische interstitielle Pneumonie (nonspecific interstitial pneumonia, NSIP), gelegentlich auch als gewöhnliche interstitielle Pneumonie (usual interstitial pneumonitis, UIP), als kryptogene organisierende Pneumonie (cryptogenic organizing pneumomia, COP), als lymphozytäre interstitielle Pneumonie (lymphocytic interstitial pneumonie, LIP) oder als akute interstitielle Pneumonie (acute interstitial pneumonia, AIP). Bei der NSIP unterscheidet man einen fibrotischen von einem zellulären Subtyp, der sich durch eine stärker ausgeprägte entzündliche Komponente auszeichnet. Bei der UIP dominiert die Fibrose. Dementsprechend findet man in der HR-CT eine meist diffuse retikuläre Verschattung bis hin zum Honigwabenmuster (honeycombing)mit allenfalls kleinen Herden einer Milchglasverschattung. Im Gegensatz dazu zeichnet sich die NSIP computertomografisch durch eine variable Kombination aus Milchglasverschattung und retikulärer Verschattung aus. Den unterschiedlichen histopathologischen Formen der ILD kommt eine wichtige prognostische Bedeutung zu. Anders als die UIP, ist die NSIP eher einer therapeutischen Intervention zugänglich und zeichnet sich durch eine bessere Prognose aus. Die interstitiellen Lungenveränderungen bei Kollagenosen sind nicht nur hinsichtlich histologischer Erscheinung heterogen; Häufigkeit des Auftretens, der klinische Verlauf und die Ausprägung der Lungenerkrankung werden nicht unwesentlich von der zugrunde liegenden Kollagenose bestimmt.
Therapeutische Strategien
Systemischer Lupus erythematodes (SLE)
Die akute Lupus-Pneumonitis ist eine seltene, lebensbedrohliche Manifestation des SLE; sie kommt in 1–10 % der Patienten vor und ist mit einer Mortalität von 50 % behaftet. Fieber, Husten, Dyspnoe, atemabhängige Thoraxschmerzen und Infiltrate, vor allem in den basalen Lungenabschnitten im konventionellen Thoraxröntgenbild sind typische Erscheinungen. In der HR-CT finden sich Milchglasverschattungen. Das histologische Bild zeichnet sich durch einen akuten Alveolarschaden aus (alveoläre Blutung, Ödem, hyaline Membranen, Immunglobulin- und Komplementablagerungen). Die schlechte Prognose erfordert eine rasche therapeutische Intervention in Form der hochdosierten i. v. Kortisonstoßtherapie (bis zu 1 g Methylprednisolon an 3 aufeinander folgenden Tagen). Darüber hinaus ist eine Cyclophosphamid-Bolus-Therapie zu erwägen. Eine ILD, vereinbar mit dem Bild der NSIP, UIP, seltener der COP oder LIP tritt in etwa 10 % der SLE-Patienten auf. Von einer therapeutischen Intervention profitieren vor allem Patienten, bei denen die entzündliche Komponente im Vordergrund steht (NSIP, Milchglasverschattungen in der HR-CT). Die Therapieentscheidung hat individuell zu erfolgen; sie orientiert sich an der Ausprägung und der Progression der Veränderungen. In schweren Fällen besteht die Therapie aus einer Kombination von Kortison (Prednisolon, initial bis zu 1 mg/kg KG) und i. v. Cyclophosphamid-Bolus-Therapie über 6 Monate, gefolgt von Azathioprin oder Mycophenolat. In weniger schweren Fällen kann Azathioprin oder Mycophenolat in Kombination mit niedrig dosiertem Kortison bereits am Beginn eingesetzt werden.
Primäres Sjögren-Syndrom (PSS)
Beim PSS wird eine ILD in bis zu 25 % beobachtet. Das Risiko einer ILD ist bei Patienten mit extraglandulären Manifestationen der Erkrankung höher als bei Patienten mit ausschließlich glandulärer Beteiligung. Die ILD tritt meist nach mehrjährigem Krankheitsverlauf auf; die häufigsten Formen sind NSIP und LIP, seltener COP oder UIP. Typische Symptome sind Husten und Belastungsdyspnoe. Differenzialdiagnostisch ist eine PAH zu erwägen. In der HR-CT findet man neben Milchglasverschattungen und retikulären Verschattungen häufiger Bronchiektasien als Folge der chronischen Entzündung der bronchialen Drüsen. Die LIP ist zusätzlich durch dünnwandige Zysten, zentrilobuläre und subpleurale Knötchen und Bronchialwandverdickungen charakterisiert. Ähnlich wie beim SLE orientiert sich die therapeutische Strategie an den Symptomen und der Progredienz der Veränderungen. Zur Behandlung der PSS-assoziierten NSIP wurde Azathioprin in Kombination mit Kortison erfolgreich angewendet. Patienten mit dem Bild einer LIP sind meist symptomatisch, eine Kortison-Therapie ist in den meisten Fällen erfolgreich.
Systemische Sklerose (SSc)
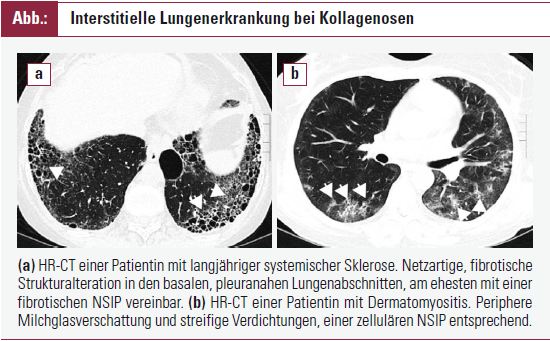
Die ILD ist eine schwerwiegende und häufige Komplikation der SSc. Bei der diffusen kutanen SSc tritt sie in bis zu 80 % der Patienten bereits in frühen Erkrankungsstadien auf. Die ILD bestimmt ganz wesentlich die Prognose der SSc; ausgedehnte Veränderungen in der HR-CT sind mit einer höheren Mortalität verknüpft. Milchglasverschattungen sprechen für eine aktiven entzündlichen Prozess (zelluläre NSIP), während streifige Verdichtungen und ein Honigwabenmuster ohne Milchglasverschattungen für eine Fibrose (fibrotische NSIP, UIP) sprechen. Randomisierte klinische Studien belegen einen geringen, jedoch nachweisbar günstigen Effekt einer Cyclophosphamid- Therapie, kombiniert mit niedrig dosiertem Kortison in frühen Stadien der Lungenerkrankung. Cyclophosphamid wird als monatliche Bolus-Therapie über zumindest 6 Monate verabreicht. Die initiale Dosis orientiert sich an der Körperoberfläche und wird dem Alter und der Nierenfunktion angepasst; die folgende Dosis wird unter Berücksichtigung des Leukozyten-Nadir festgelegt. Für Patienten, denen Cyclophosphamid nicht verabreicht werden kann, kommt als Alternative Azathioprin in Kombination mit niedrig dosiertem Kortison in Betracht.
Mischkollagenose (MCTD)
Etwa 30–50 % der Patienten mit einer Mischkollagenose entwickeln eine ILD, die mit einer Verminderung der DLCO und typischen Veränderungen in der HR-CT einhergeht und meist die basalen und peripheren Lungenabschnitte betrifft. Die therapeutischen Optionen sind ähnlich wie bei der SSc-assoziierten ILD.
Dermatomyositis/Polymyositis (DM/PM)
Eine ILD ist eine seltene Manifestation der malignomassoziierten Myositis, tritt jedoch häufiger bei der entzündlichen Myositis auf, die mit Anti-Jo-1-Antikörpern einhergeht. Das his tologische Muster entspricht meist der NSIP, seltener der UIP, COP oder AIP. Differenzialdiagnostisch sind Infektionen, die medikamentös induzierte Pneumonie (Methotrexat) und pulmonale Komplikationen, bedingt durch muskuläre Schwäche (Aspiration), zu erwägen. Das therapeutische Konzept orientiert sich wieder an der Ausprägung und Progression der Lungenveränderungen. Kortison alleine oder in Kombination mit Cyclophosphamid, Azathioprin, Mycophenolat oder auch Kalzineurininhibitoren (Tacrolimus, Cyclosporin) kommt in Betracht. Hochdosierte Immunglobuline und Rituximab wurden erfolgreich zur Behandlung der entzündlichen Myositis eingesetzt. Der Stellenwert dieser Therapieoptionen für die DM/PM-assoziierte ILD bleibt vorerst unklar.