





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Disease platforms als Grundlage integrativer Analyse in der nephrologischen Forschung
30. März 2012
Mit der Publikation des humanen Genoms 2003 wurde der Term ‚Omics‘ breit in der molekularbiologischen Forschung eingeführt: Die Bestimmung eines umfassenden Satzes an molekularen Entitäten in parallelen (multigeplexten) Assays1. Als prototypisches Beispiel sei Transcriptomics (Bestimmung der Konzentration von RNA Fragmenten) angeführt, mit den ersten Arbeiten im Hochdurchsatzverfahren in den 1990ern publiziert und mittlerweile auch zu einer Reihe an Fragestellungen in der nephrologischen Forschung angewandt2–4.
Die treibende Kraft dieser Entwicklungen war hauptsächlich auf der technologischen Seite eine Kombination von Miniaturisierung der Assay-Plattformen (bead arrays) und entsprechenden Ausleseverfahren (vor allem optische Systeme und Detektoren) und führte zur raschen Etablierung der zentralen Omics-Disziplinen Genomics, Transcriptomics, Proteomics und Metabolomics. Diese Ansätze finden in den letzten Jahren auch in translationaler klinischer Forschung breite Anwendung und erweitern unsere Kenntnis der molekularen Ausprägung von klinischen Phänotypen in der nephrologischen Forschung5–8, vor allem auch in der Kombination mit hypothesengetriebenen Ansätzen.
Hypothesengetriebene Forschung und Explorative Ansätze
Omics-Technologien versuchen die ‚Gesamtheit‘ von molekularen Entitäten eines bestimmten Typs zu bestimmen, und durch Vorliegen dieser breiten Datenlage wird die Grundlage von explorativen Analyseansätzen geschaffen: Das experimentelle Design verlangt keine Einschränkung auf z. B. einen bestimmten molekularen Prozess (oder weiter eingeschränkt auf ein bestimmtes Zielmolekül); die Gesamtheit wird bestimmt, und diese wird dann auf relevante molekulare Entitäten und Prozesse analysiert. Dieser Ansatz ist zumindest konzeptuell ansprechend, sieht aber in der Praxis weiterhin einige Limitierungen. Als Beispiel seien wieder Expressionsarrays genannt: Diese Technologie bestimmt im Regelfall die (in Abhängigkeit vom spezifischen experimentellen Design relative oder absolute) Konzentration eines definierten Satzes an Transkripten von proteinkodierenden Genen. Laut der ENSEMBL-Datenbank meint dies eine Größenordnung von 20.000 Sequenzen, und unter Verwendung von Oligochips der wesentlichen Hersteller wird aus diesem Satz an Sequenzen auch ein großer Teil erfasst. Spezifische Chips erlauben des weiteren eine Auflösung der relativen Konzentration von splice variants und dementsprechend einen Rückschluss auf die Konzentration von Proteinisoformen. Dieser Auszug an Sequenzinformation bildet jedoch nur einen Teil von Transkripten ab. Eine Vielzahl an weiteren genomischen Leserahmen wird in Transkripte übersetzt, und diese scheinen wie an Beispielen bereits gezeigt eine wesentliche regulatorische Funktion zu übernehmen (microRNA, lncRNA, etc.)6. Sohin bestimmen expression arrays einen breiten Satz von Transkripten, aber keinesfalls die Gesamtheit9. Vergleichbare Problematiken sind auch bei anderen Omics tracks zu sehen, aber hier sind wesentliche Entwicklungen im Gang, um sich der Gesamtheit weiter anzunähern. Am Beispiel Transcriptomics sei RNAseq (i. e. die explizite Sequenzierung der Gesamtheit der vorliegenden RNA aus einem bestimmten Sample) genannt, da diese Technologie hypothesenfrei sämtliche protein coding und noncoding Sequenzen erfasst und quantifiziert.
Ein weiterer Punkt ist zu berücksichtigen: das Transkriptom wovon? Am Beispiel einer Nierenbiopsie stellt sich die Auflösung des Transkriptoms zumindest nach Kompartimenten (konzeptuell nach Zelltypen), und oft repräsentiert ein gewonnenes Transkriptom die Umfassende der spezifischen Expression der Kompartimente und Zelltypen. Auch hier kann Technologie in der Probenvorbereitung (z. B. laser capture microdissection) helfen spezifischere Transkriptprofile abzuleiten10. Weitere zu berücksichtigende Punkte sind die Probengewinnung und Vorbereitung von Omics-Experimenten sowie eine adäquate Fallzahlplanung, um eine niedrige falsch-positiv Rate bei entsprechender Power in der statistischen Analyse zu erzielen11. Unter Einhaltung eines entsprechenden experimentellen Designs und Verwendung adäquater Proben(zahl), und unter Berücksichtigung der gegebenen Einschränkungen wie oben ausgeführt, erlauben Omics-Technologien jedenfalls eine effiziente und hypothesenfreie Erfassung und Quantifizierung eines großen Spektrums molekularer Entitäten.
Statistische Analyse und bioinformatische Auswertung
Eine Vielzahl von spezifischen statistischen Analyseverfahren steht zur Auswertung von Omics-Daten zur Verfügung. Wesentliches Merkmal ist die Behandlung der „curse of dimensionality“, wie am Beispiel von Transkriptomics evident12: 20.000 Hypothesen (im Falle eines gene chips mit 20.000 Proben), formuliert als „die mittlere Expression des Gens BCL-2 ist in beiden Armen einer Case-control-Gruppe gleich“ verlangen nach einem statistischen Test, um die gegebene Nullhypothese unter Ansatz eines p-Wertes (typischerweise p < 0,05) abzulehnen, wobei die Anzahl an Proben in den beiden Gruppen in der Größenordnung von 50–100 liegt. Konsequenterweise muss das statistische Verfahren Korrekturen für multiples Testen anwenden, um die falsch-positiv-Rate in definierten Schranken zu halten. Für kontinuierliche Parameter (Konzentration von Transkripten, Proteinen, Metaboliten) sind die statistischen Ansätze vergleichbar und unterscheiden sich in den spezifischen Ausprägungen in Abhängigkeit von den Charakteristika der gegebenen Omics-Daten. Aussage der Verfahren ist, inwieweit die Konzentration einer molekularen Spezies mit einer gegebenen Fehlerrate einen Unterschied zwischen Case- und Control-Gruppe aufweist. Dichotome Parameter (z. B. Vorliegen eines single nucleotide polymorphism, SNP, auf genomischer Ebene) werden beispielsweise über Kontingenztafeln ausgewertet, um die odds ratio eines SNP bezüglich Assoziation zu Case oder Control zu bestimmen.
In aller Regel bilden die (Omics-track-spezifischen) statistischen Analyseverfahren die Grundlage für weiterführende Analysen, wobei hier zwei grundsätzlich verschiedene Wege beschritten werden: Assoziation von relevanten molekularen Features mit Beschreibern des klinischen Phänotyps oder outcome, oder biologische Interpretation der (aus statistischer Sicht als relevant) identifizierten molekularen Entitäten, i.e. Anwendung von Verfahren aus der (klinischen) Statistik zusammen mit Verfahren der Bioinformatik. Gleich der spezifischen Ausprägung von statistischen Verfahren, gekoppelt mit weiter gesteigerter methodologischer Heterogenität, versuchen Omics-track-spezifische bioinformatische Verfahren die Interpretation von Resultaten aus der statistischen Analyse von Omics-Experimenten zu unterstützen13. Dies beginnt in einem ersten Schritt mit der korrekten Annotation von molekularen Spezies und umfasst unter anderem: Charakterisierung des genomischen Lokus von SNP (kodierende Regionen, Promoters, funktionale SNP, etc.), Identifikation von genomischer Region und mögliche Isoformen zu proteinkodierenden mRNA, Abschätzung von Bindungspartnern zu noncoding RNA, Unterstützung der Identifikation von Proteinen basierend auf Peptid-Fingerprints aus massenspektrometriebasierter Proteomics, Zuordnung von Metaboliten zu enzymatischen Prozessen etc. Diese bioinformatischen Methoden sind nicht per se hypothesenbildend, sondern nur aufgerufen, die gewonnene Datenlandschaft in eine Form zu überführen die im Kontext der biologischen Modelle interpretierbar ist.
Integrative Bioinformatik
Integrative Analyse, i. e. die Betrachtung von Resultaten im Kontext vorliegender Information, ist freilich das Standardverfahren wissenschaftlicher Interpretation. Eine eher methodologische als konzeptuelle Erweiterung des Integrationsgedankens resultierte wiederum aus den verfügbaren (und rasch weiter anwachsenden) Datenmengen aus den Hochdurchsatzverfahren: Aus der statistischen Auswertung von Omics-Profiles, und den aufbereitenden Maßnahmen durch bioinformatische Verfahren liegen Assoziationen von molekularen Entitäten auf den verschiedenen Ebenen molekularer Organisation (in signifikantem Ausmaß auch in der public domain) vor – und diese (zusammen mit einer selbst gewonnenen molekularen Datenlandschaft) gilt es nun zu integrieren. Idee dieses Zuganges ist es, aus dem rein deskriptiven Datenbestand auf Grund seiner Verschneidung verbesserte biologische Hypothesen abzuleiten, als aus der Betrachtung eines einzelnen Layers und/oder eines eingeschränkten Datenbestandes erzielbar wäre14. Zentrales technologisches Element in diesen Bestrebungen ist es, einen „gemeinsamen Nenner“ der molekularen Entitäten zu definieren und dann die in verschiedenen Omics-tracks gewonnen Ergebnisse zu einem gegebenen klinischen Phenotyp auf dieser gemeinsamen Basis zu annotieren. Hier wird üblicherweise das biologische (Gen-zentrierte) Standartmodell verwendet (Abb. 1).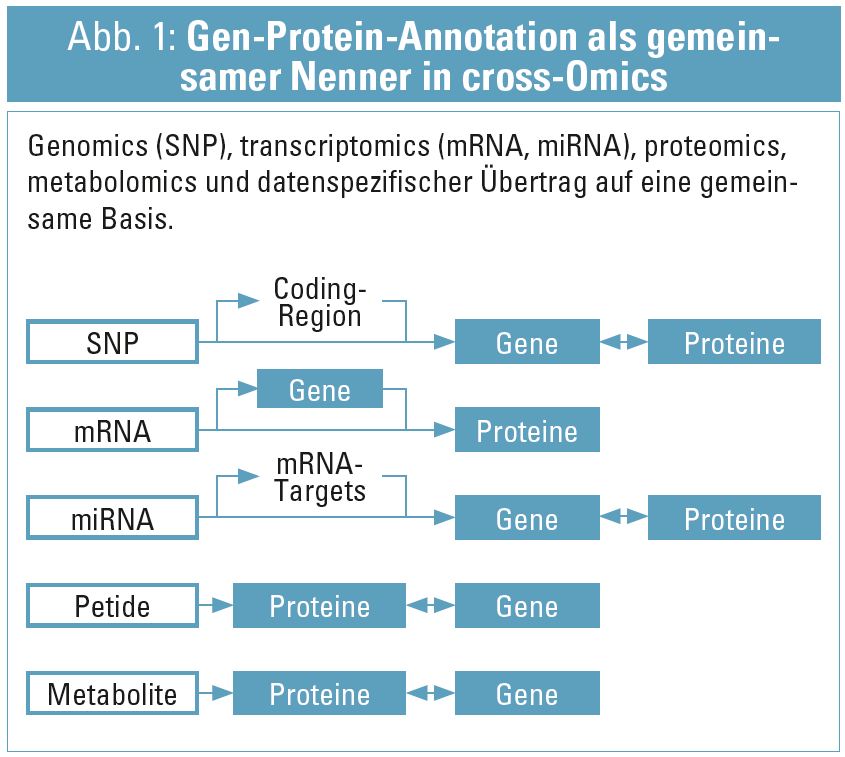
Eine Datenintegration wie hier beschrieben ist auch speziesübergreifend möglich: Daten von In-vitro- oder In-vivo- Modellen können äquivalent auf der beschriebenen Basis konsolidiert werden und brauchen vorangestellte Bioinformatik, um die Zuordnung auf den „gemeinsamen Nenner“, i. e. das humane, genzentrierte Annotationsnetz, sicherzustellen. Resultat dieser Methodik ist die Erstellung korrekter biologischer Kontextannotation unter Einbezug einer breiten Datenbasis, und erlaubt in weiterer Folge z. B. die Analyse der Konsequenz einer geänderten mRNA-Konzentration auf die Konzentration zugehöriger Proteine (so Transkriptom- und Proteomdaten vorliegen). Auch an dieser Stelle bietet die Bioinformatik nur weitere Unterstützung einer anfolgenden Interpretation – aber per se weiterhin keine Hypothesenbildung, da an dieser Stelle noch keine biologischen Modelle gebildet wurden.
Deskriptive biologische Modelle und Systembiologie
Grundsätzlich braucht biologische Modellbildung zwei Komponenten: 1. Die molekularen Entitäten und deren direkten Assoziationen (wie Gen-zu-Transkript-zu-Protein, siehe Abb. 1 und 2. deren mechanistischen/funktionalen Relationen (Protein X bindet an Protein Y im Zuge eines Signaltransduktionsweges). Hier können zum Teil wiederum statistische Verfahren herangezogen werden, um derartige Assoziationen abzuleiten. Ein Beispiel aus Transkriptomics wäre die inverse Korrelation der Konzentration von miRNA und mRNA mit dem biologischen Bild von Komplexierung und anfolgendem Abbau von mRNA durch miRNA und dementsprechend gegenläufiger Konzentrationsprofile. Neben statistischen Verfahren kann wiederum die Bioinformatik auf ein weites Spektrum von bekannten Relationen zwischen molekularen Entitäten zugreifen15, klassische Beispiele sind: direkte Proteininteraktion (z. B. Bildung eines Bcl-2-Homodimers), prozedurale Interaktionen (z. B. das Regelwerk des Apoptosesignaltransduktionsweges), und einige weitere sowie deren übergreifende Integration in Hybridnetzwerken16. Derartige Interaktionsnetzwerke können nun verwendet werden, um die annotierte Omics-Datenlandschaft zu einem gegebenen Phänotyp zu interpretieren. In Abhängigkeit von Art und Annotation des verwendeten Interaktionsnetzwerkes bietet dieser Ansatz direkte Ableitung von Hypothesen: Interaktionsnetzwerke können nach ‚betroffenen‘ Prozessen gezielt abgesucht werden, und die Ableitung dieser Erkenntnis fußt auf der konsolidierten Einarbeitung von integrierten Omics-Daten: Zuerst werden die Gen-Proteinknoten des Netzwerkes mit den Ergebnissen der (cross) Omics Analysen gekennzeichnet und anfolgend auf Ebene der Netzwerke selbst im Rahmen von vorliegenden funktionalen Gruppen (Prozesse, Pathways) untersucht. Auch vor- und nachgelagerte Prozesse können auf diesem Wege evident werden, wie beispielsweise die molekulare Charakterisierung der Schädigung in der Niere (Transkriptomics aus Biopsie) und eine Konsequenz daraus auf die renokardiale Achse (Proteomics aus Plasma), oder das damit assoziierte Proteinmuster gemessen im Harn17. Wesentlich in diesem Zusammenhang sei festzuhalten, dass diese Resultate weiterhin eine deskriptive Beschreibung der biologischen Prozesse und deren kausalen Verknüpfungen darstellen, aber bereits die grundlegenden molekularen Module von Relevanz (in einer gegebenen Ontologie abhängig vom verwendeten Netzwerktyp) erkennbar machen und somit per se zur Hypothesenbildung beitragen können: Prozess X, gekoppelt an Prozess Y, identifiziert durch die Kombination von signifikant differenzialregulierten Transcripten und zugehörigen Proteinen, erscheint relevant im Kontext des gegebenen Phänotyps, und wie stehen diese Ergebnisse im Kontext zu bereits bekannten Hypothesen?
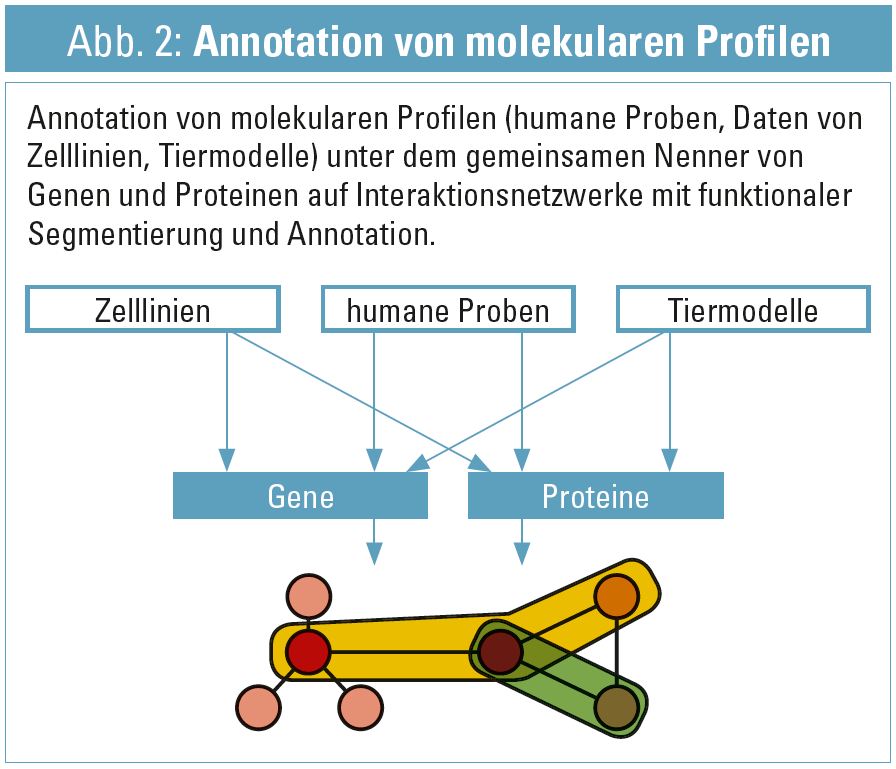 Unter Berücksichtigung all dieser Elemente sind bereits einige Voraussetzungen einer systembiologischen Herangehensweise implementiert: Integration von systemweiten Omics-Daten, Berücksichtigung von cross-Omics (von Genomik bis zu Metaboliten), speziesübergreifende Datenkonsolidierung, in ihrer Gesamtheit unter Zuhilfenahme von Interaktionsnetzwerken in molekularen Kontext gesetzt, um auch die funktionalen Zusammenhänge der molekularen Entitäten in die Modellbildung einzubauen. Ein wesentliches Element aus der spezifischen Definition der Systembiologie fehlt jedoch: Die Möglichkeit, die zeitabhängige Dynamik des beschriebenen Systems aus experimenteller sowie computergestützter Modellbildungssicht zu beschreiben (und somit direkt in einen kausalen Zusammenhang zu setzen). Dieser Teil der systembiologischen Betrachtung ist gegenwärtig nur für sehr eingeschränkte Prozesse möglich, da breite Omics-Technologien kaum in der Lage sind, Daten mit einer adäquaten (den molekularen Prozessen entsprechenden) Zeitauflösung zu liefern. Aus diesem Hintegrund ist die Systembiologie, mit einigen ersten Beispielen im Kontext der Nephrologie18, 19, weiterhin hauptsächlich als integrative Bioinformatik zu verstehen: Multi-level-Datenintegration aus den wesentlichen Omics-Disziplinen zu humanen Proben sowie Daten aus Zellkultur- und Tiermodellen, weiter kombiniert mit dem Spektrum an vorliegenden klinischen Daten.
Unter Berücksichtigung all dieser Elemente sind bereits einige Voraussetzungen einer systembiologischen Herangehensweise implementiert: Integration von systemweiten Omics-Daten, Berücksichtigung von cross-Omics (von Genomik bis zu Metaboliten), speziesübergreifende Datenkonsolidierung, in ihrer Gesamtheit unter Zuhilfenahme von Interaktionsnetzwerken in molekularen Kontext gesetzt, um auch die funktionalen Zusammenhänge der molekularen Entitäten in die Modellbildung einzubauen. Ein wesentliches Element aus der spezifischen Definition der Systembiologie fehlt jedoch: Die Möglichkeit, die zeitabhängige Dynamik des beschriebenen Systems aus experimenteller sowie computergestützter Modellbildungssicht zu beschreiben (und somit direkt in einen kausalen Zusammenhang zu setzen). Dieser Teil der systembiologischen Betrachtung ist gegenwärtig nur für sehr eingeschränkte Prozesse möglich, da breite Omics-Technologien kaum in der Lage sind, Daten mit einer adäquaten (den molekularen Prozessen entsprechenden) Zeitauflösung zu liefern. Aus diesem Hintegrund ist die Systembiologie, mit einigen ersten Beispielen im Kontext der Nephrologie18, 19, weiterhin hauptsächlich als integrative Bioinformatik zu verstehen: Multi-level-Datenintegration aus den wesentlichen Omics-Disziplinen zu humanen Proben sowie Daten aus Zellkultur- und Tiermodellen, weiter kombiniert mit dem Spektrum an vorliegenden klinischen Daten.
Aufbau von Disease Platforms
Die nephrologische Forschung ist aus dem vorliegenden Datenhintergrund, aber auch aus dem Erwachsen von Konzepten zu systembiologischer Datenanalyse bereit, integrativen Analysen hinsichtlich eines besseren Verständnisses der entsprechenden klinischen Phänotypen Raum zu geben. Das Fundament für derartige integrative Analysen wie oben beschrieben – liegt jedoch nur spärlich vor: Datenbanken mit strukturierter Haltung der Fülle an bereits verfügbaren Omics-Datensätzen, erweitert mit der Haltung von Probenidentifikation und Verlinkung zu detailierten klinischen Beschreibern der Proben selbst, und zwar umfassend für Zellkultursysteme, Tiermodelle, humane Proben und den verschiedenen Omics-tracks, weiterem Einbezug von funktionalen Studien zu spezifischen pathophysiologischen Prozessen bis hin zu Validierungsergebnissen zu spezifischen Biomarkern aber auch zu molekularen Prozessen im Zusammenhang mit spezifischen Therapien. Eine wesentliche Plattform, in der spezifische Elemente dieser Datenlandschaft der Nephrologie bereits eine strukturierte Integration erfahren, ist die „nephromine“-Datenbank (www.nephromine.org). Fokus dieser Ressource sind Transcriptomics-Daten im Kontext spezifischer klinischer Daten (die Verfügbarkeit von spezifischen klinischen Daten für die korrekte Integration von Daten aus verschiedenen Quellen und anfolgender Interpretation von Ergebnissen ist in diesem Kontext essentiell). Eine weitere Initiative ist die „KUPKB“-Datenbank (www.kupkb.org) mit dem Ziel, multi-Omics-Daten zu Nierenerkrankungen konsolidiert für integrative Analysen zur Verfügung zu stellen.
 Auf Grundlage der Heterogenität dieser zu berücksichtigenden Daten sowie auf Grund der vorliegenden Verteiltheit der Daten in der nephrologischen Forschungscommunity, sind flexible Datenmanagementkonzepte und entsprechende Softwarerealisierungen notwendig20. Die wesentlichen Objekte einer entsprechenden Integrationsplattform umfassen die Proben und deren Beschreiber (klinische Parameter, Details zu Tiermodellen etc.), die daran gekoppelten Omics-tracks und deren Resultatdaten (Profiles idealerweise auch Rohdaten) sowie die gemeinsame Annotationsbasis auf Ebene molekularer Entitäten. Neben der strukturierten Ablage dieser Datenobjekte sind auch deren Relationen zu erfassen: Welche Proben wurden im Rahmen welches Omics-tracks analysiert? War die Zuteilung in cases/controls (oder alternative Designs), gekoppelt mit der Zuteilung der probenspezifischen klinischen Daten? Diese Strukturierung erlaubt den Aufbau eines Teils des „Datengraphen“ von disease platforms: Proben mit zugehöriger Annotation (klinische Charakteristik, Details zum Tiermodell etc.), verbunden mit zugehörigen Omicstracks, daraus resultierenden Resultaten und deren gemeinsame molekulare Annotation.
Auf Grundlage der Heterogenität dieser zu berücksichtigenden Daten sowie auf Grund der vorliegenden Verteiltheit der Daten in der nephrologischen Forschungscommunity, sind flexible Datenmanagementkonzepte und entsprechende Softwarerealisierungen notwendig20. Die wesentlichen Objekte einer entsprechenden Integrationsplattform umfassen die Proben und deren Beschreiber (klinische Parameter, Details zu Tiermodellen etc.), die daran gekoppelten Omics-tracks und deren Resultatdaten (Profiles idealerweise auch Rohdaten) sowie die gemeinsame Annotationsbasis auf Ebene molekularer Entitäten. Neben der strukturierten Ablage dieser Datenobjekte sind auch deren Relationen zu erfassen: Welche Proben wurden im Rahmen welches Omics-tracks analysiert? War die Zuteilung in cases/controls (oder alternative Designs), gekoppelt mit der Zuteilung der probenspezifischen klinischen Daten? Diese Strukturierung erlaubt den Aufbau eines Teils des „Datengraphen“ von disease platforms: Proben mit zugehöriger Annotation (klinische Charakteristik, Details zum Tiermodell etc.), verbunden mit zugehörigen Omicstracks, daraus resultierenden Resultaten und deren gemeinsame molekulare Annotation.
Dieser Datenaufbau erlaubt die direkte Verschneidung mit dem 2. Teil des Datengraphen: molekulare Interaktionsnetzwerke (Abb. 2) – und hier erfolgt die direkte Verlinkung auf Ebene der gleichen molekularen Annotation.
Zentraler Nutzen derartiger Plattformen:
- Umfassende Sicht auf vorliegende Studien
- Detailierte Kenntnis des Setups und Designs von spezifischen Omics (und anderen) Studien, und daraus
- die Möglichkeit, die effektive Vergleichbarkeit von Studien zu evaluieren (Probenkollektive, Endpunkte), um daraus
- aus Sicht des Phänotyps und der Endpunkte vergleichbare Studien für integrative Analysen auszuwählen, um dann
- integrative Analysen (cross-Omics, cross-Spezies) unter vollem Einbezug der Probenbeschreiber auf Ebene von Interaktionsnetzwerken durchzuführen
Erst eine Strukturierung der Daten in dieser Form bietet die Grundlage für integrative Analysen21, – als einige Beispiele seien genannt:
- Vergleich und komplementäre Analysen zu gegebenen Proben/gegebenem Phänotyp, daraus
- die Möglichkeit, die molekulare Basis zu einem gegebenen Phänotyp genauer zu beschreiben, und damit
- in einem ersten Schritt deskriptive molekulare Modelle abzuleiten – diese bieten in weiterer Folge
- die In-silico-Evaluierung von gegebenen Biomarkerkandidaten die Ableitung von neuen Kandidaten, um in weiterer Folge
- eine Verschneidung mit gegebenen Therapieregimen und somit neue Ansätze im Umfeld von Systems medicine/Stratifizierung/Personalisierung auf systematischrationaler Grundlage voranzutreiben.
Umsetzung der Integrationsstrategie in SysKid und Ausblick
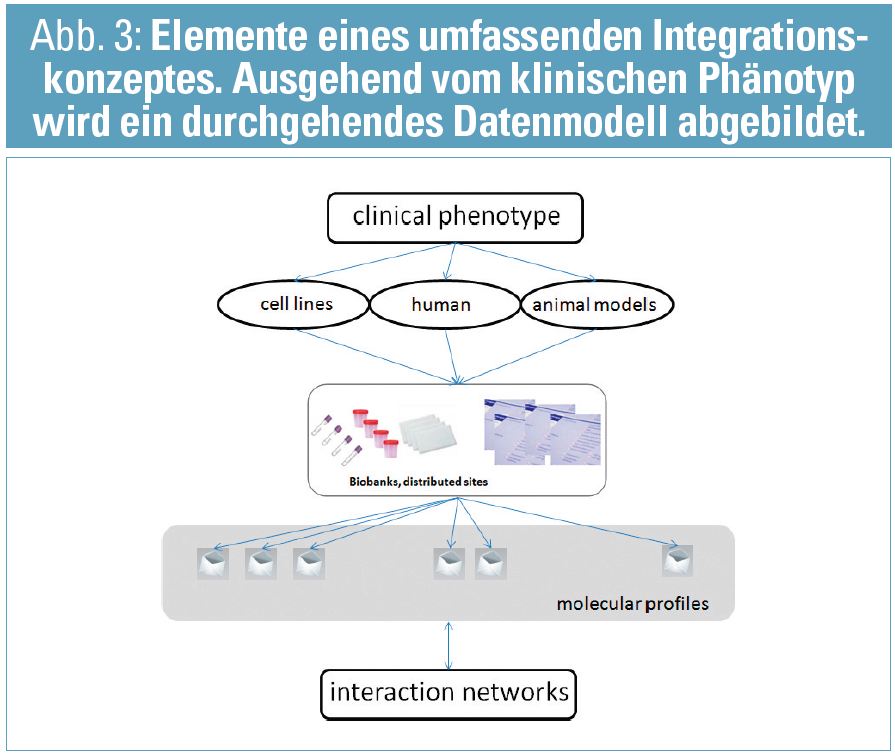
Das EU-FP7-Projekt SysKid entwickelt und verwendet diese Integrationsstrategie mit Fokus auf den frühen Status der chronischen Nierenerkrankung. Wesentliche molekulare Datenkomponenten in SysKid sind Omics-Profile (SNP, mRNA-/miRNA-transcriptomics, -proteomics, -metabolomics), gekoppelt an probenspezifische klinische Daten, ergänzt um Zellkultur und Tiermodelle und verbunden mit der Identifikation von klinischen Risikofaktoren unter Berücksichtigung der epidemiologischen Rahmenbedingungen der chronischen Nierenerkrankung (Abb. 3). Diese umfassende, multidisziplinäre Herangehensweise erbringt die notwendige Datenlandschaft, um ein Integrationskonzept – wie in diesem Artikel vorgestellt – umzusetzen und somit auch einen wesentlichen methodologischen Beitrag im Rahmen von phänotypspezifischen disease platforms in der nephrologischen Forschung zu erbringen.
1 „ome sweet“ omics – a genealogical treasure of words. J. Lederberg, A.T. McCray; The Scientist 15, 7-8 (2001).
2 Genome wide gene expression patterns of donor kidney biopsies distinguish primary allograft function. P. Hauser, Ch. Schwarz, Ch. Mitterbauer, H. M. Regele, F. Mühlbacher, G. Mayer, P. Perco, B. Mayer, T. W. Meyer, R. Oberbauer; Journal of Laboratory Investigation 84, 353-361 (2004).
3 Gene expression profiles of human proximal tubular epithelial cells in proteinuric nephropathies. M. Rudnicki, S. Eder, P. Perco, J. Enrich, K. Schreiber, Ch. Koppelstätter, G. Schratzberger, B. Mayer, R. Oberbauer, T. W. Meyer, G. Mayer; Kidney International 71, 325-335 (2007).
4 Histogenomics: Association of gene expression patterns with histological parameters in kidney biopsies. P. Perco, A. Kainz, J. Wilflingseder, A. Soleiman, B. Mayer, R. Oberbauer; Transplantation 87, 290-295 (2008).
5 Multiple New Loci Associated with Kidney Function and Chronic Kidney Disease: The CKDGen consortium. A. Köttgen et al., Nature Genetics 42, 376-384 (2010).
6 The Long and Short MicroRNAs in the Kidney. J. Ho, J.A. Kreidberg, Journal of the American Society of Nephrology 23, 400-404 (2012).
7 Urinary proteomics in the assessment of chronic kidney disease. W. Mullen, C. Delles, H. Mischak; Current Opinion in Nephrology and Hypertension 20, 654-661 (2011).
8 Metabolomic analysis of human plasma from haemodialysis patients. E. Sato, M. Kohno, M. Yamamoto, T. Fujisawa, K. Fuhiwara, N. Tanaka; European Journal of Clinical Investigation 41, 241-255 (2010).
9 The Use and Abuse of –Omes. S.J. Prohaska, P.F. Stadler, in ‘Bioinformatics for Omics data: Methods and Protocols’, B. Mayer (ed.), Humana Press (2011).
10 Hypoxia response and VEGF-A expression in human proximal tubular epithelial cells in stable and progressive renal disease. M. Rudnicki, P. Perco, J. Enrich, S. Eder, D. Heininger, A. Bernthaler, M. Wiesinger, R. Sarkozi, S. Noppert, H. Schramek, B. Mayer, R. Oberbauer, G. Mayer; Laboratory Investigation 89, 337-346 (2009).
11 Omics-Bioinformatics in the Context of Clinical Data. G. Mayer, G. Heinze, H. Mischak, M.E. Hellemons, H.J. Lambers Heerspink, S.J.L. Bakker, D. de Zeeuw, M. Haiduk, P. Rossing, R. Oberbauer, in ‘Bioinformatics for Omics data: Methods and Protocols’, B. Mayer (ed.), Humana Press (2011).
12 Statistical Analysis Principles for Omics Data. D. Dunkler, F. Sanchez-Cabo, G. Heinze; in ‘Bioinformatics for Omics data: Methods and Protocols’, B. Mayer (ed.), Humana Press (2011).
13 Transforming omics data into context: Bioinformatics on genomics and proteomics raw data. P. Perco, R. Rapberger, Ch. Siehs, A. Lukas, R. Oberbauer, G. Mayer, B. Mayer; Electrophoresis 27, 2659-2675 (2006).
14 Mapping of molecular pathways, biomarkers and drug targets for diabetic nephropathy. R. Fechete, A. Heinzel, P. Perco, K. Moenks, J. Soellner, G. Stelzer, S. Eder, D. Lancet, R. Oberbauer, G. Mayer, B. Mayer; Proteomics Clinical Applications 5, 354-366 (2011).
15 Computational reconstruction of protein interaction networks. K. Moenks, I. Muehlberger, A. Bernthaler, R. Fechete, P. Perco, R. Freund, A. Lukas, B. Mayer; in ‘Applied Statistics for Network Biology: Methods in Systems Biology’, Dehmer/Emmert-Streib/Graber/ Salvador (ed.), Wiley-VCH, page 155-178 (2011).
16 A dependency graph approach for the analysis of differential gene expression profiles. A. Bernthaler, I. Muehlberger, R. Fechete, P. Perco, A. Lukas, B. Mayer; Molecular Biosystems 5, 1720-1731 (2009).
17 Integrative Bioinformatics Analysis of Proteins Associated with the Cardiorenal Syndrome. I. Mühlberger, K. Moenks, A. Bernthaler, C. Jandrasits, B. Mayer, G. Mayer, R. Oberbauer, P Perco; International Journal of Nephrology, Article ID 809378 (2010).
18 Systems biology: opening new avenues in clinical research. F. Molina, M. Dehmer, P. Perco, A. Graber, M. Girolami, G. Spasovski, J.P. Schanstra, A. Vlahou; Nephrology, Dialysis and Transplantation 25, 1015-1018.
19 Systems biology of kidney diseases. J.C.He, P.Y. Chuang, A.Ma’ayan, R. Iyengar; Kidney International 81, 22-39 (2012).
20 Data annotation and relations modeling for integrated Omics in clinical research. A. Lukas and B. Mayer; The IIOAB Journal 1, 15-23 (2010).
21 Data graphs for linking clinical phenotype and molecular feature space. A. Heinzel, R. Fechete, J. Soellner, P. Perco, G. Heinze, R. Oberbauer, G. Mayer, A. Lukas, B. Mayer; International Journal of Systems Biology and Biomedical Technologies 1, 11-25 (2012).

Ursprünglich erschienen:
Neph 01|2012
Neph 01|2012