





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Neurologische Symptome bei metabolischen Enzephalopathien
14. Dezember 2012
Organerkrankungen, Elektrolytstörungen, Malnutritionen, aber auch Medikamente können sekundär zu schweren neurologischen Erkrankungsbildern führen. Die neurologischen Symptome sind breit gefächert und spiegeln die zeitliche Dynamik, Dauer und Schwere der Organdysfunktion beziehungsweise das Ausmaß der Fehlernährung wider.
Im Folgenden werden die hepatische und die urämische Enzephalopathie, die osmotische Myelinolyse und die Wernicke-Enzephalopathie beschrieben.
Hepatische Enzephalopathie
Die hepatische Enzephalopathie (HE) tritt bei akutem Leberversagen und chronischer Lebererkrankung auf. Die Pathogenese der zerebralen Funktionsstörung ist komplex und nur zum Teil bekannt. Die Akkumulation von Ammoniak im Gehirn scheint dabei eine zentrale Rolle zu spielen. Ammoniak induziert in den Astrozyten eine Glutaminproduktion und führt über einen osmotischen Gradienten zu einer Astrozytenschwellung und Astrozytendysfunktion, eine zusätzliche Störung im Tryptophanstoffwechsel und anderen Neurotransmittern wird angenommen, und eine Stimulation des N-Methyl-D-Aspartat-Rezeptors (NMDA-Rezeptor) führt zur Nitritoxid-Freisetzung, sekundärer Vasodilatation, Hyperämie und konsekutivem Hirnödem.
Die Einteilung der HE erfolgt nach Ätiologie und klinischer Präsentation. Unterschieden wird zwischen einer HE bei akutem Leberversagen, HE bei portosystemem Bypass ohne Lebererkrankung und der HE bei Leberzirrhose. Die klinische Klassifikation beurteilt Bewusstseinslage, neuropsychologische Störungen und neurologische Symptome. Am gebräuchlichsten sind die West-Haven-Kriterien (WHC) mit 5 Schwerestufen der Erkrankung (Tab.). Klinische Verschlechterungen können sich akut innerhalb von Stunden bis Tagen, aber auch chronisch über Monate entwickeln. Kurze temporäre Verschlechterungen (episodische HE) mit dazwischen monatelangen asymptomatischen Phasen sind möglich und meist durch diätetische Fehler mit hoher Eiweißbelastung, gastrointestinale Blutungen, Infektionen, Alkoholexzess, Dehydratation und durch den Gebrauch hepatotoxischer Medikamente getriggert.

HE-Stadien: Das Stadium I der HE ist eine Leberfunktionsstörung ohne klinisch-neurologische und intellektuelle Auffälligkeiten. Zeigen sich jedoch in erweiterten psychometrischen Testverfahren neuropsychologische Störungen, spricht man von einer latenten bzw. minimalen hepatischen Enzephalopathie (MHE). Mit Fortschreiten der Erkrankung kommt es zur progredienten qualitativen und quantitativen Beeinträchtigung des Bewusstseins. Auch die Entwicklung eines Komas ist möglich und stellt damit die schwerste Form der HE dar (WHC IV). Störungen des Schlaf-Wach-Rhythmus, Verlangsamung des Denkablaufes und eine verwaschene Sprache sind frühe Begleiter der Erkrankung. Ein Tremor bei feinmotorischen Leistungen ist bereits im Stadium I und eine Asterixis („flapping tremor“, „wing beating tremor“) ab Stadium II fassbar. Eine Asterixis ist jedoch nicht pathognomonisch für eine HE und kann auch bei anderen metabolischen Erkrankungen und Intoxikationen auftreten. Ebenso häufig wird ein rigid-akinetisches Syndrom beobachtet. Fokale Herdzeichen sind nicht typisch für eine HE, da die HE ein globaler zerebraler Prozess ist. Das Auftreten fokaler Herdzeichen erfordert immer eine strukturelle Abklärung zum Ausschluss einer Zweitpathologie (z. B. intrakranielle Blutung durch Gerinnungsstörungen).
Zunehmende sozioökonomische Bedeutung gewinnt die minimale hepatische Enzephalopathie. Die Prävalenz der MHE wird bei PatientInnen mit Leberzirrhose mit 30–70 % angegeben. Die Mini-Mental State Examination (MMSE) kann nur als grober Screening-Test zum Erfassen der MHE gesehen werden und muss bei negativem Ergebnis durch andere, spezifischere Testverfahren erweitert werden (z. B. Number-Connection-Test A und B, Line-tracing-Test, Mosaic-Test, Digit-Symbol-Test). In diesen spezifischen Tests zeigen PatientInnen mit einer MHE bereits früh eine Beeinträchtigung der Aufmerksamkeit, der räumlichen Orientierung, des Kurzzeitgedächtnisses und eine Störung der frontal-exekutiven Leistungen. Studien konnten belegen, dass die MHE mit einer verminderten Lebensqualität verbunden ist, beim Lenken eines Kraftfahrzeugs eine höhere Anzahl an Fahrfehlern auftreten und das frühe Auftreten möglicherweise ein unabhängiger Prädiktor für den weiteren Verlauf der Leberzirrhose ist.
Die Diagnose einer hepatischen HE ergibt sich aus der Anamnese eines chronischen beziehungsweise akuten Leberversagens und dem klinisch-neurologischen Bild. Die üblichen Parameter wie Elektrolyte, Blutbild, Leber- und Nierenfunktionsparameter sowie Gerinnung dienen zur Einstufung der Schwere der Lebererkrankung und dem rechtzeitigen Erkennen von Komplikationen. Der Ammoniakspiegel im Serum korreliert nur bei der episodischen HE mit der Schwere der neurologischen Symptome. Bei der MHE und der persistierenden hepatischen Enzephalopathie kann von der Höhe des Ammoniakspiegels nicht auf die Schwere der neurologischen Erkrankung geschlossen werden.
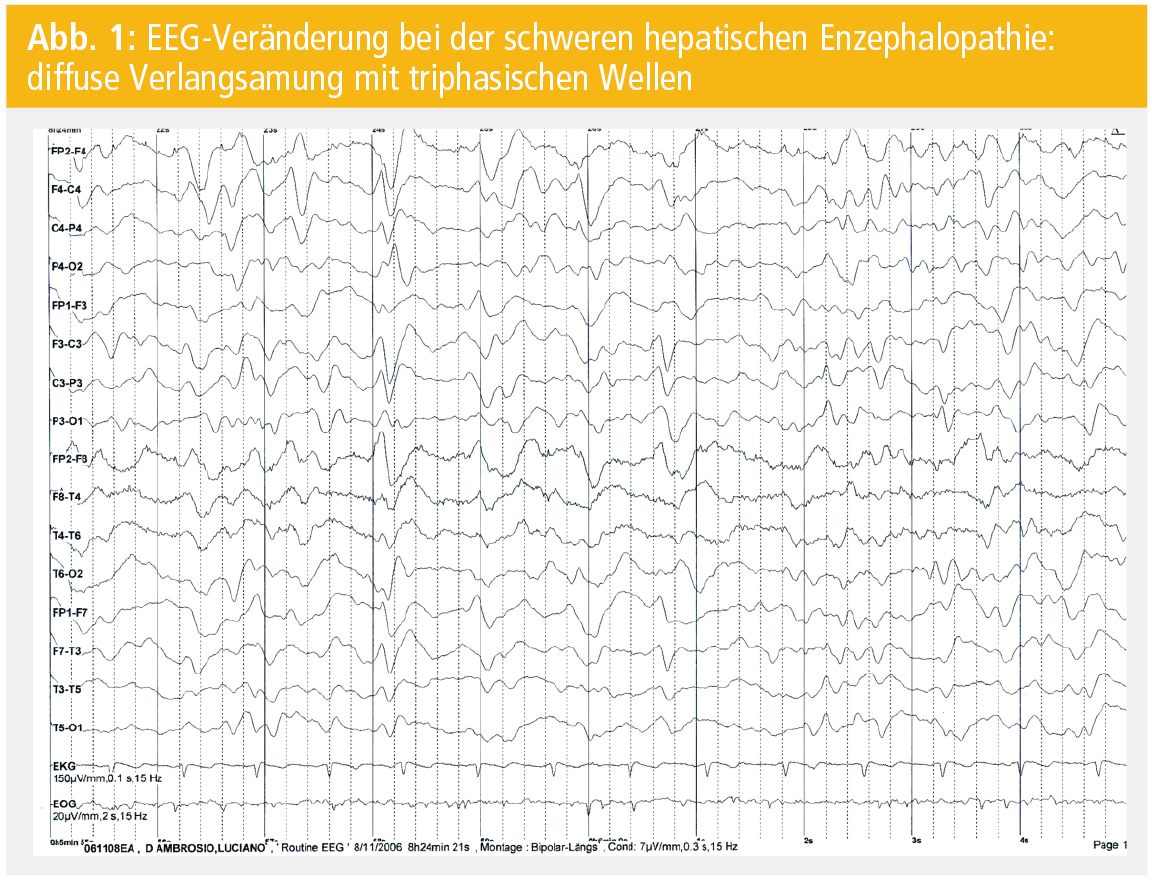
Die EEG-Ableitung zeigt in der frühen Erkrankungsphase eine diffuse Verlangsamung und im fortgeschrittenen Stadium zusätzlich triphasische Wellen (Abb. 1). Triphasische Wellen können jedoch auch bei anderen metabolischen Erkrankungen auftreten und sind daher nicht spezifisch für eine HE. Während die somatosensorisch und visuell evozierten Potenziale keine diagnostische Bedeutung in der HE haben, finden die auditorischen P-300-Wellen in der Beurteilung der neurofunktionellen Störung bei der Leberzirrhose Verwendung. Weiters scheinen erste Daten zur elektrophysiologischen Beurteilung der MHE mittels spezieller visueller Stimulation („critical flicker frequency“) Erfolg versprechend zu sein. In der Magnetresonanztomographie (MRT) können sich bei schwerer HE symmetrische, hyperintense Läsionen in der T1-Gewichtung vorwiegend im Globus pallidus, in der Capsula interna, dem Putamen, dem Nucleus caudatus und dem Mesencephalon zeigen.
Die Therapie der HE ist das Vermeiden bzw. die Therapie von allen Faktoren, welche eine Verschlechterung der hepatischen Situation bewirken (z. B. Alkoholkarenz, Vermeidung hepatotoxischer Medikamente, Therapie gastrointestinaler Blutungen, Infektbehandlung). Die Gabe von Laktulose, Probiotika, L-Ornithin-L-Aspartat und Rifaximin in Zyklen von 3–6 Monaten reduziert die Produktion und Adsorption von Ammoniak im Darm. Bei der MHE führt die Gabe von Laktulose und Rifaximin zu deutlich besseren Ergebnissen in den psychometrischen Tests.
Nichtkonvulsive epileptische Anfälle treten in 10–20 % der PatientInnen mit einer HE III/IV auf und werden mit Benzodiazepinen und Levetiracetam therapiert. Bei der fortgeschrittenen HE (West-Haven-Kriterien III/IV) und im akuten Leberversagen kommt es in bis zu 95 % der Fälle zu einer Hirndruckerhöhung. Ein kontinuierliches Hirndruck-Monitoring ist jedoch nur in Einzelfällen indiziert. Hirndruckkrisen werden mit Analgosedierung, Mannitol und hypertoner Kochsalzlösung behandelt.
Die Anlage eines portosystemen Shunts (TIPS) kann zu einer Verbesserung der HE führen und die Organtransplantation hinauszögern. Die Lebertransplantation stellt die maximale Therapie einer HE dar. Die immunsuppressive Therapie, Abstoßungsreaktionen und opportunistische Infektionen nach Organtransplantation können jedoch zu neuen neurologisch-diagnostischen und therapeutischen Herausforderungen werden (siehe unten).
Urämische Enzephalopathie
Ähnlich der HE zeigt auch die urämische Enzephalopathie (UE) ein breites Spektrum an neurologischen Symptomen. Die Pathogenese der UE ist ebenso nicht im Detail verstanden, komplex und multifaktoriell. In der Niereninsuffizienz akkumulieren verschiedenste organische Substanzen, welche als Neurotoxine mit der Freisetzung exzitatorischer und inhibitorischer Neurotransmitter interagieren. Die Urämie führt zu einem gestörten zerebralen Metabolismus, einer reduzierten Sauerstoffextraktion und einer Störung der ATPase-abhängigen Kalziumpumpe an der Hirnzelle. Der Kalziumgehalt im zerebralen Kortex von PatientInnen mit Niereninsuffizienz ist doppelt so hoch wie beim Gesunden, wobei der renale sekundäre Hyperparathyreoidismus eine zusätzliche pathogenetische Rolle spielen dürfte.
Klinik: Während beim akuten Nierenversagen die klinische Symptomatik mit der Höhe des Harnstoffs im Serum korreliert, zeigt die chronische Niereninsuffizienz trotz hohem Serumharnstoff oft nur diskrete Symptome. Müdigkeit, Apathie, Konzentrationsstörungen, feinschlägiger Aktionstremor und eine Reduktion der Aufmerksamkeitsspanne sind frühe Symptome des Nierenversagens. Bei zunehmender Erkrankung treten emotionale Labilität, Antriebsminderung, Kurzzeitgedächtnisstörung, fehlendes abstraktes und divergentes Denken, Greifschablonen, positiver Palmomental- und Schnauzreflex sowie Schlafstörungen hinzu.
Der schwere Krankheitsverlauf ist durch ein Delir, visuelle Halluzinationen, epileptische Anfälle und fluktuierende Bewusstseinslage gekennzeichnet. Ein Koma ist möglich. Eine rigide Muskeltonuserhöhung und eine Hyperreflexie sind lange im Krankheitsverlauf vorhanden und noch bei beginnendem Koma feststellbar. Eine fluktuierende Hemiparese mit wechselnder Seite tritt in bis zu 45 % auf („alternierende Hemiparese“). Myoklonien und eine Asterixis kommen erst spät im Krankheitsverlauf hinzu. Ein Meningismus ist bei bis zu 30 % der PatientInnen fassbar.
Die Dialysemöglichkeit führt zu einer deutlichen Reduktion der schweren UE. Aber auch unter der Dialysebehandlung klagen viele PatientInnen über Antriebs- und Gedächtnisstörungen. Milde kognitive Störungen sieht man bei bis zu 30 % der PatientInnen, schwere kognitive Defizite sind bei 10 % der PatientInnen fassbar. Ältere DialysepatientInnen zeigen eine deutlich höhere Inzidenz eines demenziellen Syndroms als die Vergleichsaltersgruppe. Dies wird auf das höhere vaskuläre Risikoprofil der Nierenerkrankten und der Entwicklung einer Multiinfarktdemenz zurückgeführt. Zusätzlich leiden DialysepatientInnen unter Schlaferkrankungen (z. B. obstruktives und zentrales Schlafapnoesyndrom). Die prolongierte Schlafdeprivation und die verstärkte Tagesmüdigkeit verschlechtern die kognitiven Funktionen.
Die Diagnose einer UE wird anhand der Anamnese, der erhöhten Nierenfunktionsparameter und verminderter Diuresemenge gestellt. Die zerebrale Bildgebung dient dem Ausschluss anderer struktureller, intrakranieller Pathologien, eine diffuse zerebrale Atrophie mit erweiterten Ventrikeln kommt jedoch häufig zur Darstellung. Einzelne Fallserien beschreiben eine Signalveränderung in den T2-gewichteten MRT-Aufnahmen in den Basalganglien, der periventrikulären weißen Substanz und in der Capsula interna. Diese Veränderungen sind unter Dialyse und nach Organtransplantation reversibel. Urämische PatientInnen mit Meningismus können im Liquor cerebrospinalis eine milde Pleozytose und Eiweißerhöhung (Zellzahl < 25/mm3, Protein < 100 mg/dl) haben. Ein entzündlicher Prozess muss dann jedoch stets mikrobiologisch und serologisch ausgeschlossen werden.
Die EEG-Veränderungen sind unspezifisch und bei der akuten Urämie deutlicher ausgeprägt. Es zeigt sich eine diffuse Verlangsamung im Delta/Theta-Frequenzbereich mit frontaler Akzentuierung. Im späten Krankheitsverlauf sind bilaterale Spike-Wave-Komplexe ohne klinische Anfallsaktivität in bis zu 20 % ableitbar.
Dialysekomplikationen: Ein Dysäquilibriumsyndrom ist heute eine seltene Komplikation einer Dialysebehandlung und wird vorwiegend bei PatientInnen mit einer länger bestehenden schweren Urämie und schwerem Hypertonus beobachtet. Ursache ist ein osmotischer Gradient zwischen Hirnzelle und Extrazellularraum durch eine zu rasche Dialyse. Da Harnstoff nur langsam aus der Hirnzelle diffundieren kann, führt ein zu rasches Absenken des Harnstoffs im Serum zu einem Wassereinstrom in die Hirnzelle und zur Entwicklung eines Hirnödems. Das Syndrom ist durch akut einsetzende migräniforme Kopfschmerzen, Übelkeit, Erbrechen, Muskelkrämpfe, Delir, Myoklonien und epileptische Anfälle gekennzeichnet und tritt am Ende einer Dialyse auf. Ein vorübergehend kurzes Dialyseintervall und eine geringe Dialyseaustauschmenge führen in der Regel zur raschen Stabilisierung der Symptome.
Unter der Verwendung aluminiumhältiger Dialysate und auf Aluminium basierender Phosphatbinder kam es früher zum Auftreten einer Dialyse-Enzephalopathie und Dialyse-Demenz. Das Erkrankungsbild war gekennzeichnet durch einen subakuten Verlauf einer Dysarthrie, Dysphasie, Apraxie, schwerer Persönlichkeitsveränderung, Psychose, Myoklonus und epileptische Anfälle mit tödlichem Ausgang innerhalb weniger Monate. Heute ist dies eine nur mehr sporadisch beobachtete Erkrankung und auf einen Behandlungsfehler durch Verwendung von Aluminiumsubstanzen zurückzuführen.
Die Differenzialdiagnose der UE sind die osmotische Myelinolyse, die hypertensive Enzephalopathie, ein reversibles posteriores Leukenzephalopathiesyndrom und eine Medikamentenüberdosierung durch die verzögerte renale Elimination.
Nach Organtransplantation werden unter der immunsuppressiven Therapie mit Tacrolimus und Zyklosporin schwere Enzephalopathiesyndrome beschrieben. Die Neurotoxizität dieser Substanzen ist multifaktoriell und führt zur Schwellung der Axone und Demyelinisierung. Tremor, epileptische Anfälle und Dysarthrie bis zur Anarthie sind frühe Symptome der toxischen Nebenwirkungen. Das MRT zeigt vorwiegend in den subkortikalen, occipitalen, temporalen und parietalen Hirnregionen hyperintense Veränderungen in den FLAIR- und T2-gewichteten Sequenzen. Eine Dosisreduktion der Substanzen bringt eine rasche Besserung der Symptome.
Osmotische Myelinolyse (pontine und extrapontine Myelinolyse)
Adams und MitarbeiterInnen beschrieben erstmals 1959 das Bild einer zentralen symmetrischen Demyelinisierung in der Pons („central pontine myelinolysis“, CPM). Lange wurde die Erkrankung mit chronischem Alkoholabusus in Zusammenhang gebracht und pathologische Ähnlichkeiten zum Marchiafava-Bignami-Syndrom (Demyelinisierung des Corpus callosum und anderer Kommissurenbahnen) gesehen.
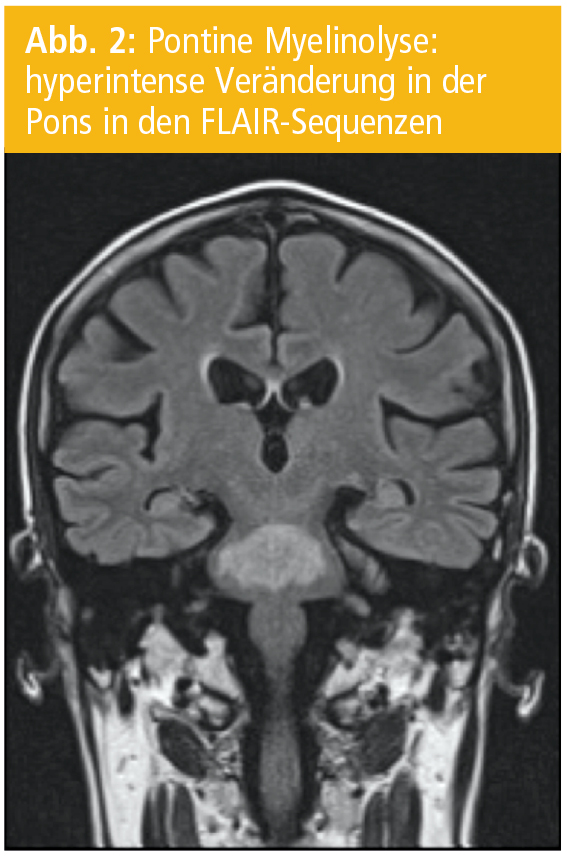
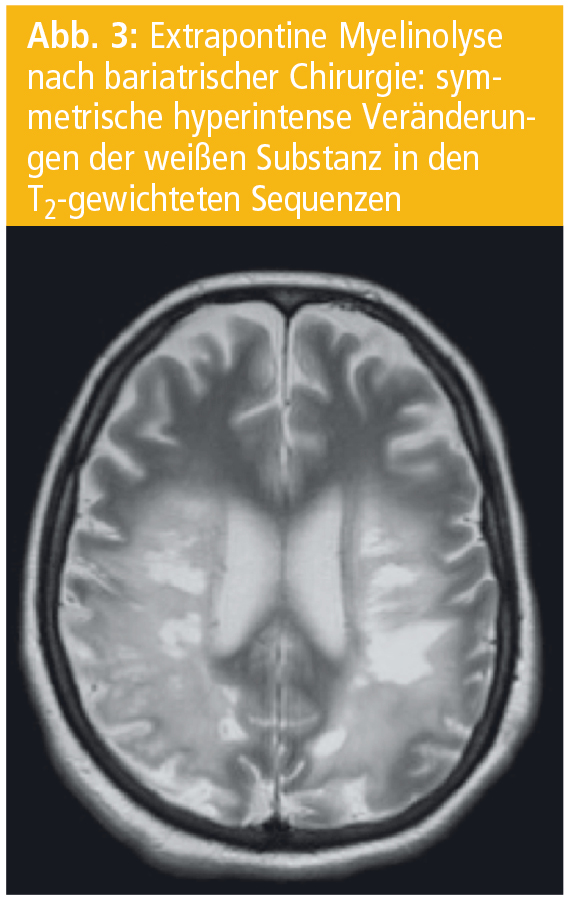
Hauptursache der CPM ist jedoch eine rasche Korrektur einer Hyponatriämie. Die Veränderungen können auch extrapontine Hirnareale wie Thalamus, Striatum, weiße Substanz des Kleinhirns und die tiefen Schichten des Neokortex mit der angrenzenden weißen Substanz betreffen („extrapontine myelinolysis“, EPM; Abb. 2). Häufig treten eine CPM und EPM gemeinsam auf. Alkoholismus in Kombination mit einer Hyponatriämie sind die häufigsten Ursachen einer CPM (Abb. 3).
Da die Erkrankung jedoch auch bei Mangelernährung, unter der Therapie mit Immunsuppressiva nach Organtransplantation (speziell Lebertransplantation), bei Malignomen, Verbrennungen und nach sportlicher Langzeitbelastung (z. B. Triathleten, Marathonläufer) auftritt, werden als weitere Faktoren extreme Blutzucker- und Plasmaproteinschwankungen, aber auch ein rascher Ausgleich einer Hypernatriämie diskutiert.
Gemeinsame pathologische Endstrecke der Schädigung ist ein osmotisches Dysäquilibrium, welches zur Endothelzellschädigung, Zusammenbruch der Blut-Hirn-Schranke und osmotischer Myelinolyse in unterschiedlichen Hirnarealen führt. Eine zusätzliche Freisetzung von myelotoxischen Substanzen durch den osmotischen Stress der Zelle ist wahrscheinlich. Die CPM präsentiert sich mit nukleären Hirnnervenausfällen, Pseudobulbärparalyse, Tetraparese und Ataxie. Bei der EPM treten extrapyramidale Bewegungsstörungen, Paresen und kognitive Störungen in den Vordergrund. Schwerste Verlaufsformen mit einem Locked-in-Syndrom bei der CPM und einer kortikalen Deafferenzierung bei der EPM sind möglich.
Die Diagnose erfolgt mittels MRT-Untersuchung. Die diffusionsgewichteten Aufnahmen zeigen frühzeitig die charakteristisch lokalisierten Veränderungen, eine diskrete Kontrastmittelaufnahme ist an den Läsionsrändern gelegentlich möglich. Evozierte Potenziale untermauern die funktionelle Störung.
Management: Eine langsame Serumnatrium-Korrektur ist zur Vermeidung einer CPM essenziell. Natriumwerte von über 120 mmol/l im Serum (SNa) mit kurzer Dauer sind nur selten mit dem Entstehen einer CML verbunden. Eine Natriumsubstitution bei SNa-Werten unter 120 mmol/l und einer Dauer über 48 Stunden weist ein höheres Risiko für eine CPM/EPM auf, da kompensatorische, intrazellulär angehäufte Osmolyte (z. B. Myo-Inositol, Taurine, Glutamin) die Zellmembran nur langsam passieren und damit das osmotische Dysäquilibrium prolongieren. Die Natriumsubstitution erfolgt mit hypertoner Kochsalzlösung. Ein Anstieg des SNa um 0,5 mmol pro Stunde darf nicht überschritten werden. Bei SNa-Werten unter 105 mmol/l kann initial, bis zum Erreichen dieses Wertes, eine raschere Substitution angestrebt werden. Der Einsatz von Vasopressin-Rezeptorantagonisten („Vaptane“) könnte in Zukunft bei der euvolämischen Hyponatriämie eine weitere Option zur Korrektur des SNa darstellen. Die SNa-Korrektur ist stets mit einer strikten und engmaschigen Überwachung der Laborwerte verbunden.
Die moderne Bildgebung und Labordiagnostik hat die Diagnose, Therapie und Prognose der CPM/EPM entscheidend verbessert. Das initiale klinische Bild und die Bildgebung lassen jedoch nicht auf das Outcome schließen. Eine Restitutio ad integrum ist trotz schwerster Symptomatik und eindrucksvoller Bildgebung möglich.
Wernicke-Enzephalopathie (Thiaminmangel-Enzephalopathie)
Thiamin (Vitamin B1) ist ein essenzielles Koenzym im Kohlehydrat- und Lipidstoffwechsel und wird zur Synthese von Aminosäuren und Neurotransmittern benötigt. Ein Thiaminmangel führt zu einer Wernicke-Enzephalopathie (WE), einem akut einsetzenden Erkrankungsbild mit neurokognitiven Verhaltensauffälligkeiten, Augenbewegungsstörungen und Gangataxie (Wernicke-Enzephalopathie-Trias).
Eine WE wurde lange in engem Zusammenhang mit chronischem Alkoholismus und begleitender Mangelernährung gesehen. Die Erkrankung tritt jedoch auch bei einseitiger Ernährung, Essstörungen, gastrointestinalen Erkrankungen und nach gastrointestinalen und bariatrischen chirurgischen Eingriffen auf.
Klinik: Der Vitamin-B1-Speicher ist nach 2–3 Wochen entleert, klinische Symptome können jedoch erst nach Wochen auftreten. Fehlende zeitliche und situative Orientierung, verzögerter Gedankenduktus und Apathie sind die wichtigsten Symptome der WE (80 %) und werden häufig missinterpretiert. Die optomotorische Störung mit horizontalem Blickrichtungsnystagmus, konjugierter Blickparese und partiellen Augenmuskelparesen tritt nur bei 30 % der Erkrankten auf, eine Rumpf- und Gangataxie kommt in 20 % vor. Die klassische Trias der WE wird somit nur bei 10 % der Erkrankten beobachtet.
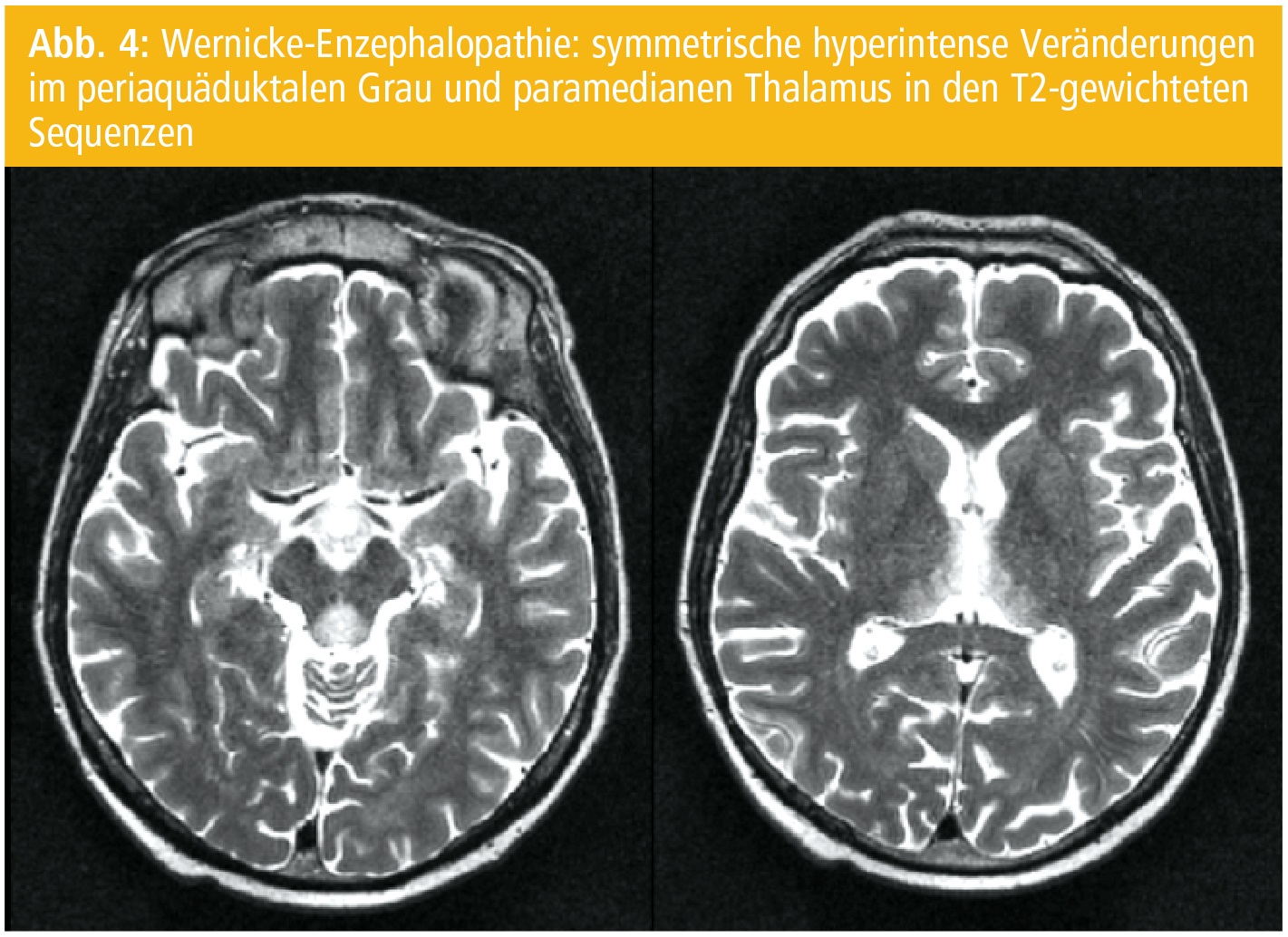
Die WE ist eine klinische Diagnose. Es gibt keine Laborparameter, die die Diagnose akut bestätigen. Das MRT zeigt in nur knapp 50 % typische symmetrische Veränderungen im periaquäduktalen Grau und im paramedianen Thalamus (Abb. 4). Hämorrhagische Veränderungen in den Läsionen sind möglich.
Management: Die WE ist ein neurologischer Notfall, und der Verdacht erfordert die umgehende intravenöse oder intramuskuläre Thiaminsubstitution. Es gibt keine Studien über optimale Dosis, Frequenz und Dauer der Therapie. Empfehlungen sind zwischen 500–1.500 mg Thiamin intravenös pro Tag für 2–3 Tage mit einer anschließenden Erhaltungsdosis von 100–250 mg Thiamin für mindestens weitere 3–5 Tage. Die Ophthalmoplegie bessert sich unter der Thiaminsubstitution meist innerhalb weniger Stunden, die Ataxie innerhalb von Tagen, eine Besserung der kognitiven Störung stellt sich innerhalb von Wochen ein. Initiale Tagesdosen von 100–250 mg Thiamin müssen als zu niedrig angegeben werden und begünstigen bleibende zerebrale Schäden.
Die Mortalität der WE wird auch heute noch mit 20 % angegeben. Der Großteil der PatientInnen überlebt in einem Korsakow-Syndrom, einem schweren amnestischen Syndrom mit fehlender Fähigkeit, Neues zu erlernen, und fehlendem divergenten Denken.
Literatur bei der Verfasserin

AutorIn: Priv.-Doz. Dr. Bettina Pfausler
Universitätsklinik für Neurologie, Neurologische Intensivstation , Medizinische Universität Innsbruck

Ursprünglich erschienen:
neuro 04|2012
neuro 04|2012

