





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Muskelerkrankungen im Kindesalter
29. Juni 2012

Klassifikation
In den letzten Jahren hat die wachsende Zahl an bekannten Genen und Genorten, die zu einer Muskelerkrankung führen können, deutlich gemacht, dass phänotypisch verschiedene Erkrankungen auf Mutationen desselben Gens beruhen (Phänotypvariabilität) und umgekehrt, dass Erkrankungen mit demselben oder überlappendem Phänotyp auf unterschiedliche Gendefekte zurückzuführen sind (Genotypvariabilität bzw. „genetic splitting“). Die Klassifikation wird derzeit noch überwiegend nach klinischen und muskelbioptischen Kriterien getroffen:
- Die Muskeldystrophien sind mit den Dystrophinopathien, den Gliedergürteldystrophien, der fazioskapulohumeralen Dystrophie (FSHD), den Emery-Dreifuss-Muskeldystrophien, den kongenitalen Dystrophien und den nur selten bereits im Kindesalter auftretenden distalen Muskeldystrophien die größte Krankheitsgruppe. Meist führt ein primärer Defekt der Muskelzelle zur Muskelfaserdegeneration und in weiterer Folge zur Fibrose mit entsprechendem histopathologischem Befund. Klinisch kommt es zu einer progressiven Muskelschwäche in variabler Geschwindigkeit und Ausprägung. Der CK-Wert ist meist deutlich erhöht und kann das 40-Fache des Normwertes erreichen.
- Die kongenitalen Strukturmyopathien stellen mit einer Inzidenz von ca. 1 : 17.000 seltene Erkrankungen dar und machen ca. 1/10 aller Myopathien im Kindesalter aus. Sie werden nach Strukturauffälligkeit der Muskelfaser, die zum Teil nur in der Elektronenmikroskopie erkennbar ist, klassifiziert: nemaline Stäbchen, Cores, „bodies“, zentralständige Kerne. Klinisch sind sie meist langsam oder gar nicht progredient. Der CK-Wert im Serum ist häufig normal oder nur leicht erhöht.
- Die myotone Dystrophie (DM1) und die proximale myotone Dystrophie (DM2) sind mit einer Inzidenz von ca. 1 : 8.000 nach der DMD die häufigsten hereditären Muskelerkrankungen. Sie stellen Multisystemerkrankungen dar, die neben einer Muskelschwäche und Myotonie charakteristischerweise auch Katarakte, Herzrhythmusstörungen und Hypogonadismus verursachen. Die CK ist leicht bis mäßig erhöht. Die Elektromyographie (EMG) bringt oft erst bei älteren PatientInnen ein typisch myotones Entladungsmuster, die Muskelbiopsiebefunde zeigen nur unspezifische Ergebnisse, weshalb die genetische Verifizierung bei Verdacht unverzichtbar ist.
- Die (muskulären) Ionenkanalerkrankungen sind sehr seltene Erkrankungen. Die häufigsten von ihnen zeigen eine Inzidenz von 1 : 23.000 (Myotonia congenita Becker) bzw. 1 : 100.000 (hypokaliämische periodische Paralyse). Es handelt sich um defekte Natrium-, Kalzium- (Einstrom oder Ausstrom) oder Chloridkanäle, die über ein hyperexzitables Sarkolemm zur Myotonie oder über ein hypoexzitables Sarkolemm zu einer Paralyse mit temporär beeinträchtigter Muskelfunktion führen. Die CK ist oft um das ca. 2-Fache erhöht. Eine EMG ist bei kleinen Kindern oft noch nicht möglich, weshalb Anamnese und Klinik wegweisend zur genetischen Diagnostik sind.
- Metabolische Myopathien haben die gestörte Energieproduktion der Skelettmuskulatur gemeinsam. Grundlage dieser Erkrankungen sind kongenitale Defekte der mitochondrialen Atmungskette, des Glykogenstoffwechsels, des Lipidstoffwechsels oder des Purinstoffwechsels. Die häufigste Störung des Glykogenstoffwechsels ist der Morbus Pompe mit einer Prävalenz von 1 : 40.000; die mitochondrialen Erkrankungen haben eine Prävalenz von 1 : 5.500 in der Gesamtbevölkerung. Diese Erkrankungsgruppe ist sehr heterogen und lässt sich von der Symptomvielfalt und den Verlaufsformen nicht zusammenfassen. Bei Symptomen wie belastungsabhängiger Muskelschwäche, Myalgien und Episoden mit Myoglobinurie sollte aber gezielt nach metabolischen Myopathien gesucht werden.
Klinik
Alle neuromuskulären Erkrankungen zeigen das Symptom Muskelschwäche. Dieses zeigt im Kindesalter altersspezifische Manifestationsformen: Säuglinge fallen als „floppy infants“ mit einer generalisierten muskulären Hypotonie auf. Sie zeigen einen niedrigen Muskeltonus („floppy“) und kaum Spontanbewegungen. Im Traktionsversuch hängt der Kopf nach hinten und im Gegensatz zu den zentral bedingten „floppy infants“ bleiben Beine und Arme schlaff. Oft leiden sie, bedingt durch die Beteiligung von Zwerchfell und/oder Interkostalmuskulatur, unter pulmonalen Komplikationen. Die Liste der Differenzialdiagnosen eines Floppy-Infant-Syndroms ist lange und beinhaltet nicht nur neuromuskuläre, sondern auch metabolische Erkrankungen, Syndrome und Störungen sowie Malformationen des Zentralnervensystems.
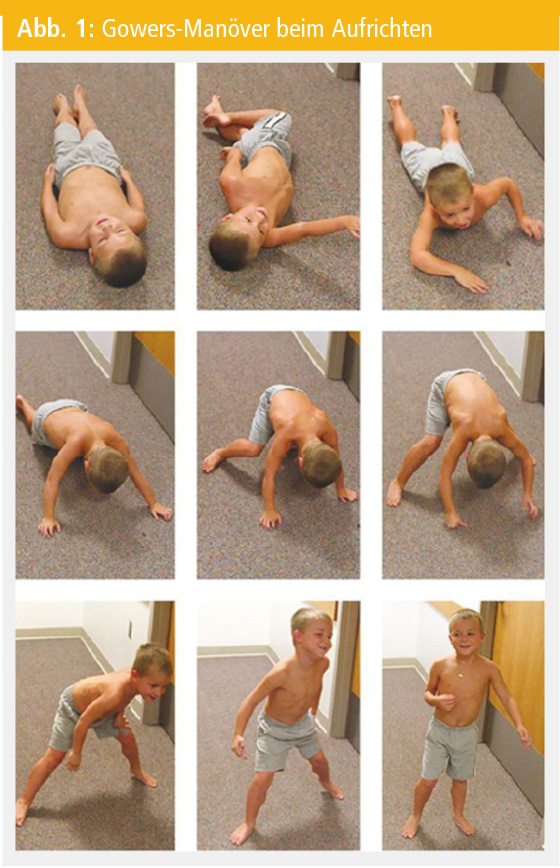
Kleinkinder mit Muskelerkrankungen fallen durch das Nichterreichen von motorischen Meilensteinen oder den Verlust bereits erworbener motorischer Fähigkeiten auf. Im Falle einer proximalen Muskelschwäche kommt es zu Schwierigkeiten beim Treppensteigen, zum positiven Gowers-Manöver beim Aufstehen vom Boden und zum Trendelenburg-Gangbild mit vermehrtem Stolpern. Bei der DMD ist der Zehenballengang ein häufiges Frühsymptom, das durch den fibrotischen Umbau und der konsekutiven Verkürzung der Wadenmuskulatur verursacht wird. Immer wieder kommt es bereits in der präsymptomatischen Phase, in der die Kinder noch eine altersgerechte Entwicklung bzw. keine wegweisenden Symptome zeigen, zu einer zufälligen Diagnosestellung. Erhöhte Transaminasen führen zu einer Bestimmung der Kreatinkinase (CK), deren Höhe auf eine Muskelerkrankung hinweisen kann.
Schulkinder und Jugendliche fallen meist durch eine zunehmende Schwäche und/oder Belastungsintoleranz auf.
Diagnostik
Anamnese: Familienanamnese, Alter bei Erkrankungsbeginn, Dynamik des Fortschreitens der Erkrankung, Verteilungsmuster der Muskelschwäche und -atrophien bzw. -pseudohypertrophien sowie zusätzliche Symptome wie Kardiomyopathie, Kontrakturen, Versteifung der Wirbelsäule (rigid spine), Myalgien oder Rhabdomyolysen machen oft Verdachtsdiagnosen möglich.
Elektrophysiologische Untersuchungen haben ihren Stellenwert bei der Identifizierung von Motoneuronenerkrankungen, Neuropathien und Myasthenien, die klinisch nicht immer eindeutig von Muskelerkrankungen abzugrenzen sind. Nicht nur die Durchführung der elektrophysiologischen Untersuchung, sondern auch deren Interpretation unterscheidet sich aber in vielen Aspekten von jenen der Erwachsenen, weshalb die elektrophysiologische Untersuchung von Kindern in pädiatrisch spezialisierten elektrophysiologischen Labors durchgeführt werden sollte.
Muskelbiopsie: Liegt nun der Verdacht auf eine Muskelerkrankung vor, ist die Muskelbiopsie meist der nächste Schritt. Der Muskelbiopsiebefund mit der Beschreibung morphologischer, immunhistochemischer und elektronenmikroskopischer Merkmale ermöglicht in vielen Fällen die Diagnosestellung bestimmter Muskelerkrankungen und im Anschluss die gezielte genetische Diagnostik. Falls die Familie keine Diagnostik durch Muskelbiopsie wünscht, sollte bei passender Klinik zumindest der Morbus Pompe (Glykogenose Typ II) mittels Bestimmung der Enzymaktivität aus Lymphozyten (besser: aus Hautfibroblasten oder Muskelzellen) ausgeschlossen werden, da bei dieser Erkrankung durch Enzymersatztherapie eine deutliche Verbesserung des klinischen Verlaufs erreicht werden kann.
Genetische Diagnostik: Manchmal ist es sinnvoll, primär einen genetischen Befund zu erheben: So kann bei einem 4-jährigen Knaben mit Wadenpseudohypertrophie und proximaler Muskelschwäche – erkennbar am positiven Gowers-Phänomen – und einer CK-Erhöhung über das 10-Fache des Normwertes die genetische Abklärung mittels MLPA zum Nachweis einer Deletion oder Duplikation im Dystrophingen erfolgen. Mit diesem Verfahren können 72 % der DMD-Patienten diagnostiziert werden. Bei weiteren 26 % bringt eine Sequenzierung des Dystrophingens den Nachweis einer Punktmutation. Led
iglich 2 % der DMD-Patienten können mit diesen Verfahren nicht diagnostiziert werden. Bei ihnen liegen andere genetische Konstellationen vor (z. B.: deep intronic point mutation). Molekulargenetische Untersuchungen als primäre Diagnostik stehen auch für die Becker-Muskeldystrophie, die myotonen Dystrophien 1 und 2, die fazioskapulohumerale Dystrophie und bei Verdacht auf Lamindefekte zur Verfügung.
Der Weg zur genetischen Diagnose ist oft kompliziert, lange und kostspielig, da oft mehrere Gene konsekutiv sequenziert werden müssen. Mit Hilfe neuer Technologien soll der Weg zur genetischen Diagnose künftig deutlich kürzer und rationaler werden. Das EU-geförderte Projekt „NMD-Chip“ (= neuromuscular disorder chip) konnte im September 2011 eine erfolgreich validierte Technologie vorweisen, mit der es gelingt, zahlreiche bekannte Gendefekte, die neuromuskuläre Erkrankungen verursachen können, binnen 72 Stunden bis einer Woche zu untersuchen. Gleichzeitig wird an Sequenzierungsverfahren gearbeitet, die unbekannte krankheitsverursachende Genveränderungen detektieren.
Auch wenn für die meisten Erkrankungen keine kurative Therapie zur Verfügung steht, ist die genetische Diagnostik von großer Bedeutung. Sie ermöglicht die Abklärung weiterer Familienmitglieder und eine genetische Beratung. Dieser Aspekt ist in der Pädiatrie von besonderer Bedeutung, da die Familienplanung oft noch nicht abgeschlossen ist. Aufgrund zunehmender Daten in der Genotyp-Phänotyp-Korrelation können Verlauf und Prognose besser abgeschätzt und notwendige Maßnahmen geplant werden. In manchen Fällen ermöglicht die positive genetische Diagnose beim Indexpatienten nicht nur die Diagnose bei weiteren asymptomatischen Familienmitgliedern, sondern in einigen Fällen, wie z. B. bei BMD- und DMD-Carrierinnen, auch deren prophylaktische Mitbehandlung bzw. die Feststellung asymptomatischer Organmanifestationen (Kardiomyopathie bei DM1, BMD und DMD-Carrierinnen). Angesichts der Fortschritte bei der Entwicklung molekularer Therapien bleibt natürlich auch zu hoffen, dass die genetische Diagnostik künftig auch entscheidend für weitere Therapieverfahren sein wird.
Medikamentöse Therapiemöglichkeiten
Morbus Pompe ist durch die Enzymersatztherapie (ERT), die seit 1998 klinisch erprobt wird und seit 2006 zur Verfügung steht, die erste therapierbare Muskelerkrankung. Der progrediente Verlauf dieser Glykogenspeichererkrankung kann durch die ERT massiv verlangsamt werden und führt zu einer deutlichen Verbesserung von Lebensqualität und Lebensdauer. Eine frühe Diagnosestellung und ein rascher Therapiebeginn sind für die Prognose entscheidend, weshalb bei suspekter Symptomatik eine sofortige Diagnosesicherung bzw. ein Ausschluss erfolgen soll. Problematisch ist die ERT vor allem bei der infantilen Verlaufsform des Morbus Pompe. Je weniger Restenzymaktivität vorhanden ist, umso eher kommt es zur Bildung von Antikörpern gegen die rekombinante humane Alpha-1,4-Glukosidase. Ob eine zusätzliche Immunmodulation eine Verbesserung der Ansprechrate auf die ERT bringt, ist derzeit noch unklar und wird im Rahmen von klinischen Studien untersucht.
Bei DMD sind Glukokortikoide bislang die einzige nachgewiesen wirksame Substanz zur Verlangsamung des Kraftverlustes. Der Wirkmechanismus ist noch nicht im Detail geklärt, wird aber größtenteils der antiinflammatorischen Wirkung zugeschrieben, nachdem muskelzellnekrosengetriggerte lokale Entzündungsprozesse einen aggravierenden Effekt auf den pathologischen Prozess haben dürften. Es kommt nachweislich zu einer Verlängerung der Gehfähigkeit um durchschnittlich 2 Jahre; positive Effekte auf die zu erwartende Entwicklung einer Skoliose, der respiratorischen Funktion und der Kardiomyopathie sind beschrieben. Es gilt daher als Goldstandard in der Plateauphase, in der keine motorischen Fortschritte mehr gemacht werden, aber auch noch keine Rückschritte zu erkennen sind, um das 4. Lebensjahr (LJ) die Steroidtherapie zu initiieren und auch nach Verlust der Gehfähigkeit um das 10. LJ fortzusetzen, solange keine relevanten Nebenwirkungen auftreten. Im Jahr 2012 soll nun eine große multizentrische Studie zum Vergleich der Wirksamkeit und Nebenwirkungsprofile von Prednison (0,75 mg täglich oder 10 Tage on/10 Tage off intermittierend) und Deflazacort (0,9 mg täglich) begonnen werden.
Standards of Care
Abgesehen von der Steroidtherapie lassen sich die Standards of Care für die DMD auch auf zahlreiche andere Muskelerkrankungen übertragen. Spätestens nach erfolgter Diagnosestellung sollten Kinder mit Muskelerkrankungen an ein pädiatrisches neuromuskuläres Zentrum angebunden und dort regelmäßig (meist halbjährlich) kontrolliert werden. So können diagnostische und therapeutische Maßnahmen rechtzeitig geplant und begonnen werden. Neben der medizinischen und funktionell therapeutischen Betreuung der betroffenen Kinder sollte auch stets eine psychosoziale Beratung und Begleitung für den/die Patienten/-in und seine/ihre Familie angeboten werden, da die Qualität der Krankheitsbewältigung wesentlich für die Lebensqualität und die Compliance ist. Kontaktmöglichkeiten zu Selbsthilfegruppen sollten ebenso zum Angebot gehören.
Die Geh-, Steh- und Sitzfähigkeit sollte möglichst lange erhalten bleiben. Maßnahmen, die dazu beitragen können, sind Dehnungsübungen zur Vermeidung von Kontrakturen und Physiotherapie, Versorgung mit Orthesen, Stehständer und Mieder bzw. orthopädische Operationen. Nach Verlust der Gehfähigkeit besteht das Risiko einer immobilitätsbedingten Osteoporose, das durch eine Steroidtherapie verstärkt wird, weshalb regelmäßige Kontrollen der Vitamin-D-Versorgung und evtl. auch der Knochendichte notwendig sind.
Stabiliserung der Wirbelsäule: Bei Auftreten einer progredienten Skoliose sollte nach Gehverlust eine frühzeitige Stabilisierung der Wirbelsäule erfolgen. Sie kann die Sitzfähigkeit erhalten, dient zur Prophylaxe lagerungsbedingter Schmerzen und mildert respiratorische Komplikationen. Es ist erwiesen, dass die Stabilisierung der Wirbelsäule ein wesentlicher Faktor ist, um die Lebensdauer zu verlängern und die Lebensqualität langfristig zu verbessern. Eine späte Operation hat ein deutlich höheres kardiorespiratorisches Risiko, die Korrektur der Skoliose ist meist nur eingeschränkt möglich. Diese Fakten müssen wiederholt mit den Familien besprochen werden, da der Eingriff an der Wirbelsäule stark angstbesetzt ist und viel Aufklärungs- und Bedenkzeit benötigt.

BiPAP-Therapie: Genauso verhält es sich mit dem rechtzeitigen Beginn einer nächtlichen nichtinvasiven Atemunterstützung mittels BiPAP. Da bei vielen Muskelerkrankungen die Atemmuskulatur beteiligt ist, kommt es zu einer (progredienten) restriktiven Ventilationsstörung. Ab einer Vitalkapazität < 60 % (spätestens 40 %) bzw. bei PatientInnen, die aufgrund ihres Alters bzw. Entwicklungsstandes keine Lungenfunktionsprüfung durchführen können, sollte eine Polysomnographie durchgeführt werden. Die respiratorische Insuffizienz wird zunächst im Schlaf manifest und führt zu Symptomen wie morgendliche Kopfschmerzen, Konzentrationsstörungen, Tagesmüdigkeit und depressiver Stimmungslage. Durch Beginn der BiPAP-Therapie ab dem Stadium der nächtlichen hyperkapnischen Hypoventilation kommt es zu einer Besserung dieser Symptome, Exazerbationen der akuten respiratorischen Insuffizienz können hintan gehalten werden.
Die intermittierende BiPAP-Therapie ist prinzipiell auch bei kleinen Kindern und Säuglingen möglich, bedarf aber häufig einer individuell angepassten Zielsetzung und ausreichend Zeit und Geduld, da die Masken oft speziell angefertigt werden müssen und die Geräteeinstellung nur langsam schrittweise optimiert werden kann. Neben der nächtlichen BiPAP-Therapie zählen auch die Komplettierung des Impfstatus, die Optimierung der Ernährungssituation (gegebenenfalls via PEG-Sonde), die Atemtherapie und das Sekretmanagement zur Prophylaxe und Behandlung der respiratorischen Komplikationen.
Dieses Management hat in den letzten Jahrzehnten zu einer deutlichen Verbesserung der Lebensqualität und Verlängerung der Lebensdauer geführt.
Die Verlängerung der Lebenserwartung hat allerdings zur höheren Relevanz der kardialen Beteiligung als lebenslimitierendem Faktor geführt. Bei der DMD ist bei ersten echokardiographischen Anzeichen einer Kardiomyopathie der Einsatz von ACE-Hemmern und/oder Betablockern indiziert. Ob der frühzeitige Einsatz der medikamentösen Therapie ab dem 10. Lebensjahr einen Vorteil bringt, wird derzeit in einer multizentrischen Studie in Deutschland geklärt.
Die steigende Lebenserwartung führt auch dazu, dass zunehmend eine Transition zur Erwachsenenmedizin stattfinden muss. Bei der Betreuung im multidisziplinären Team, bestehend aus OrthopädInnen, PulmonologInnen, KardiologInnen, funktionellen TherapeutInnen, ErnährungsberaterInnen, SozialarbeiterInnen und PsychologInnen, nehmen die NeurologInnen eine zentrale und koordinierende Stellung ein.
Netzwerke und Ausblick
Das EU-geförderte europäische Netzwerk TREAT-NMD (Translational Research in Europe for the Assessment and Treatment of Neuromuscular Disease) wurde 2007 gegründet und verbindet Europas führende SpezialistInnen auf dem Gebiet der neuromuskulären Erkrankungen in Kooperation mit weltweiten PatientInnenorganisationen. Es hat das Ziel, die Behandlung von neuromuskulär erkrankten Menschen zu verbessern und zu vereinheitlichen und somit die Therapieforschung zu beschleunigen. TREAT-NMD betreibt mehrere Projekte zu den Themen „Standards für Diagnose und Therapie“ und „Patientenregister für DMD, spinale Muskelatrophie, FKRPopathien und kongenitale Muskeldystrophien“.
PatientInnenregister ermöglichen (künftig) Genotyp-Phänotyp-Korrelationen, Aussagen über regionale Versorgungsstrukturen und das rasche und gezielte Finden von PatientInnen, die für ein bestimmtes Studiendesign in Frage kommen. Das „Care and Trial Site Registry“ dient ebenfalls dem Ziel, die Standards of Care zu implementieren und ermöglicht eine einfachere und raschere Durchführung von internationalen multizentrischen klinischen Studien. Im Jänner 2012 waren 274 Zentren aus 46 Ländern registriert, wobei derzeit das Gottfried-von-Preyer’sche Kinderspital in Wien das einzige registrierte Zentrum in Österreich ist.
Klinische Studien: Durch TREAT-NMD kam es in den letzten Jahren zu einer stark zunehmenden Zahl an klinischen Studien. Hatte man früher das Problem, überhaupt eine qualitativ hochwertige Studie zustande zu bringen, befinden wir uns heute in der glücklichen Situation, dass Studienthemen und -zeitpunkte genau ausgesucht und geplant werden müssen.
Diese (Phase-IIb-/III-)Studien betreffen einerseits bekannte Medikamente mit neuem Einsatzgebiet (z. B.: Idebenon) oder neue molekulare Therapien, wie beispielsweise das Exon-Skipping. Diese Methode wird aktuell in einer großen multizentrischen Phase-III-Studie am Exon 51 des DMD-Gens getestet. Man hofft damit den Verlauf der DMD bei der milderen Dystrophinopathie Becker-Muskeldystrophie abmildern zu können. Mittels Anti-Sense-Oligonukleotiden können während des Transkriptionsprozesses jene Exons übersprungen werden, die zu Stopcodons führen. Dadurch bleibt das Leseraster erhalten, und es wird ein kürzeres, aber funktionstüchtiges Dystrophineiweiß in der Muskelzelle gebildet, das bei der DMD sonst fehlt. Derzeit ist aber noch unklar, ob die Menge des gebildeten Dystrophins ausreicht, um den Krankheitsverlauf merkbar beeinflussen zu können, und ob der Körper das neu gebildete Eiweiß langfristig annimmt. Aussagen bezüglich der Wirksamkeit bleiben noch abzuwarten. Die Methode des Exon-Skippings wird derzeit an der DMD getestet, da hier eine ausreichende Zahl an Patienten für Phase-III-Studien rekrutiert werden können. Wenn sich die Methode bewährt, könnte sie auch auf andere Exons des Dystrophingens und auch anderer krankheitsverursachender Gene angewendet werden, weshalb sie ein großer Hoffnungsträger für viele seltene, genetisch bedingte Erkrankungen darstellt.
Weiterführende Literatur:
– Bertino E et al., Congenital muscular dystrophies: a brief review. Semin Pediatr Neurol 2011; 18:277–288
– Bushby K et al., Diagnosis and Management of Duchenne Muscular Dystrophy. Part 1: Diagnosis, pharmacological and psychosocial management. Lancet Neurol 2010; 9:77–93
– Bushby K et al., Diagnosis and Management of Duchenne Muscular Dystrophy. Part 2: Implementation of multidisciplinary care. Lancet Neurol 2010; 9:177–189
– Cirak S et al., Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 2011; 378:595–605
– Goemans M et al., Systemic Administration of PRO051 in Duchenne’s Muscular Dystrophy. New Engl J Med 2011, 364:1513–22
– Lutz S et al., Kongenitale Strukturmyopathien. Medizinische Genetik 2009; 21:316–321
– Manzur AY et al., Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev 2008; (1):CD003725
– Muntoni F, Voit T, The congenital dystrophies in 2004: a century of exciting progress. Neuromuscul Disord 2004; 14:635–649

Ursprünglich erschienen:
neuro 02|2012
neuro 02|2012

