





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Neurologische Symptome im Rahmen internistisch-immunologischer Krankheitsbilder
14. Dezember 2012

Symptome des zentralen und peripheren Nervensystems im Rahmen internistisch-immunologischer Erkrankungen sind ein derart weites, Buchbände füllendes, multiple organspezifische Disziplinen involvierendes Gebiet, sodass sich dieser Artikel im Allgemeinen auf die Übersicht der diagnostischen Abklärung, der klinisch-neurologischen Manifestationen und der therapeutischen Vorgehensweisen bei zentralnervösen Manifestationen internistischer (oder besser: systemischer) immunologischer Krankheitsbilder beschränkt.
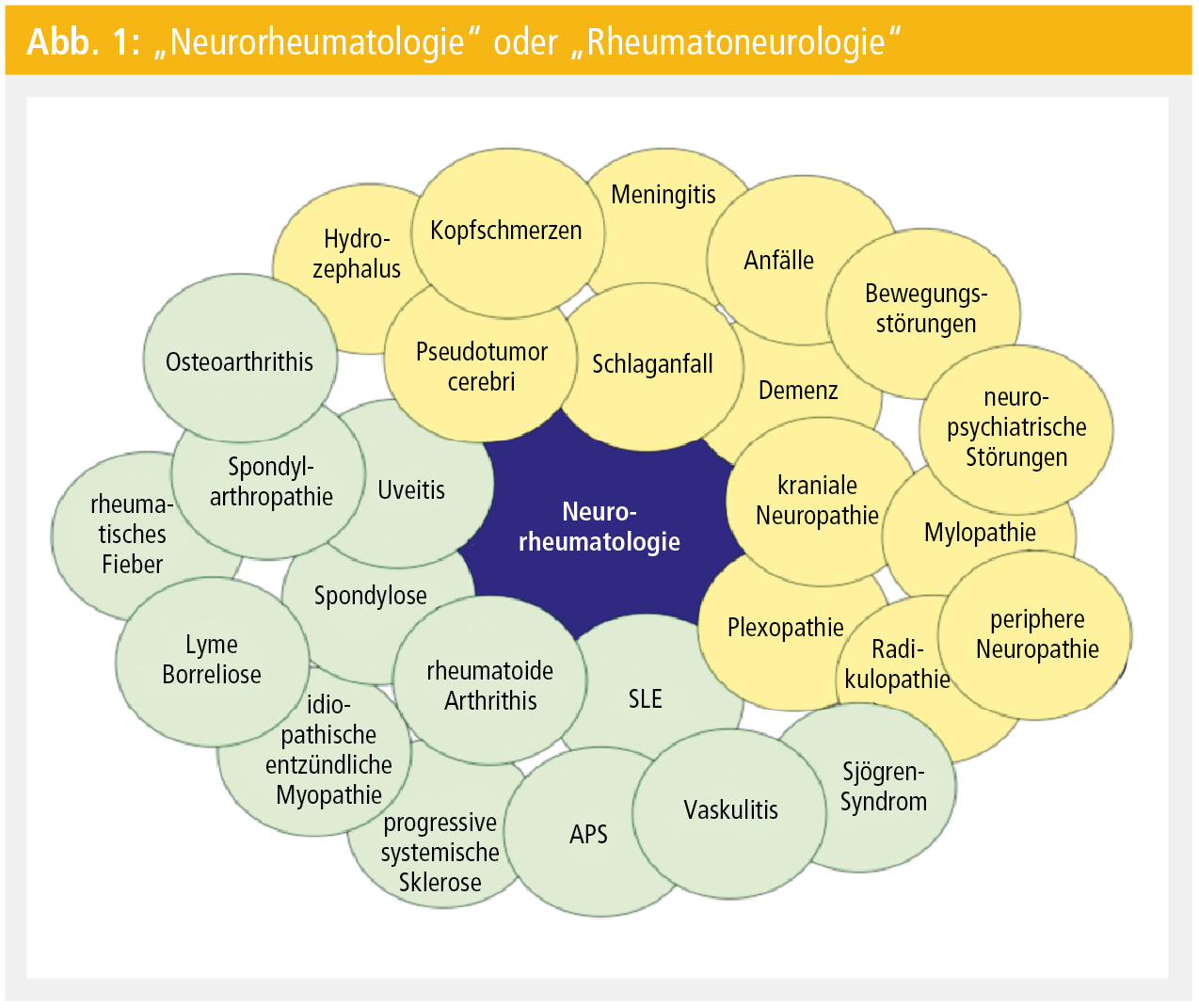
Nahezu jede (auto-)immunologisch bedingte Erkrankung kann konkomitant mit einer abzugrenzenden neuroimmunologischen Erkrankung (beispielsweise Multiple Sklerose und Mb. Crohn, rheumatoide Arthritis, Mb. Bechterew, Hashimoto-Thyreoiditis, juveniler Diabetes mellitus) oder mit neurologischen Manifestationen (beispielsweise der Multiple Sklerose-ähnliche Leukenzephalopathie bei Zöliakie, paraneoplastische Syndrome) als primäre oder sekundäre Folge der zugrunde liegenden Systemerkrankung einhergehen. Dieser Artikel fokussiert daher auch nur auf Systemerkrankungen, die zum Formenkreis rheumatologischer und vaskulitischer Erkrankungen gehören. Aus der Vielfalt auch dieser „neuro-rheumatologischen“ (oder „rheumato-neurologischen“) Erkrankungen (Abb. 1) werden einige wenige Krankheitsbilder aber ausführlicher dargestellt, beispielgebend für immunologisch (vaskulitisch) bedingte Systemerkrankungen, die sich mitunter als neurologische Erstmanifestation präsentieren.
Diagnostik
Anamnese und neurologische Untersuchung
Für Neurologen/Neurologinnen ist natürlich das Erkennen und Erfassen einer neurologischen Symptomatik genuin immanent, aber der Verdacht auf eine immunologisch bedingte Systemerkrankung verdichtet sich bei folgenden anamnestischen oder aktuellen klinischen Hinweisen:
- Hinweise auf Systemerkrankung:
- Fieber unklarer Genese
- andere Organbeteiligungen
- Hinweise auf organspezifische Symptome: beispielsweise
- Hautläsionen (ischämisch, inflammatorisch, hämorrhagisch)
- Augenbeteiligung (Uveitis)
- Nierenbeteiligung (Glomerulonephritis)
- Hinweise auf multiple/ungewöhnliche ischämische Ereignisse: beispielsweise
- multiple Infarkte in unterschiedlichen Organen innerhalb kurzer Zeitabstände
- ischämische Ereignisse bei jungen Erwachsenen
- Spontanaborte
Für Nichtneurologen/Nichtneurologinnen ist es hingegen von Bedeutung, zu wissen, dass die diagnostizierte immunologische Systemerkrankung auch zentralnervöse und/oder periphere neurologische Symptome und Erkrankungen verursachen kann.
Labordiagnostik
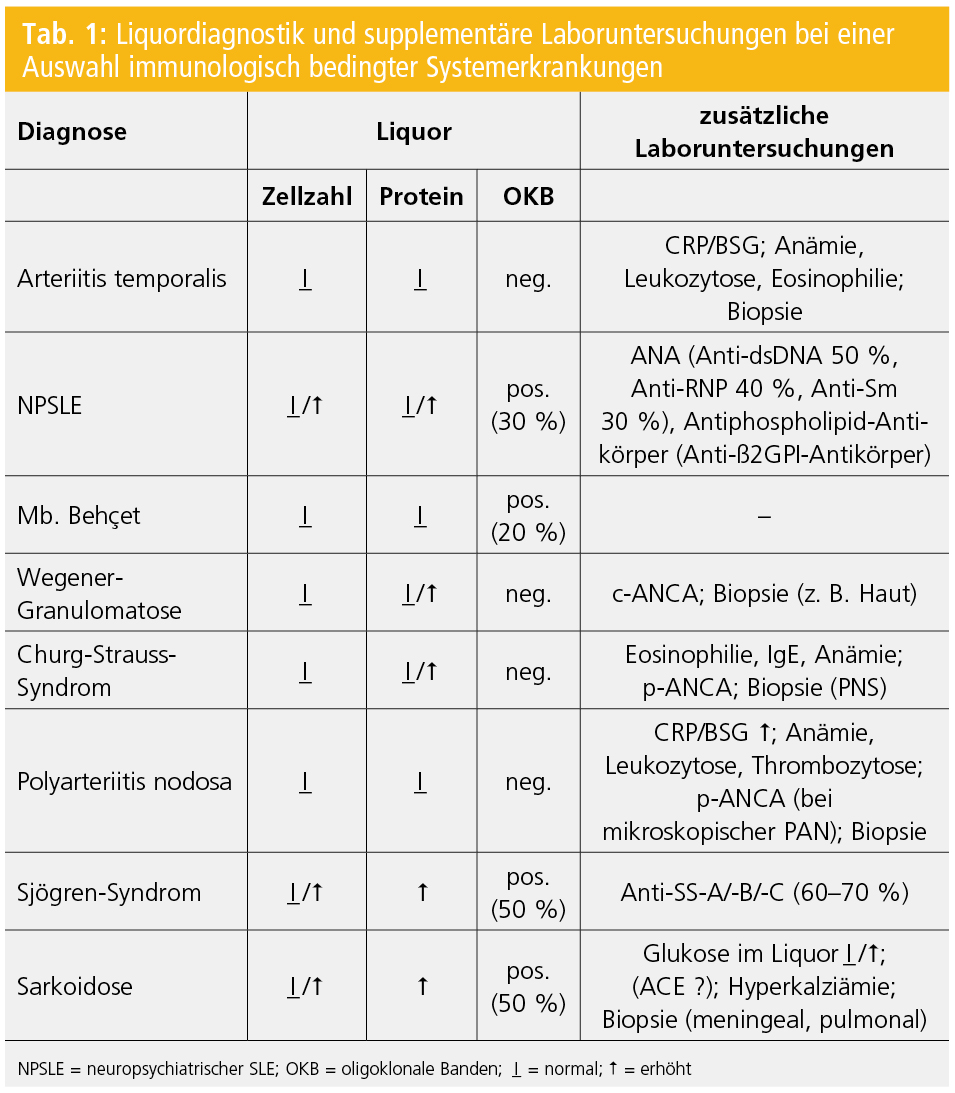
Gegenwärtig gibt es keinen spezifischen Biomarker oder Laborparameter zur Diagnostik einer präklinischen oder klinisch-manifesten neurologischen Symptomatik im Rahmen einer Vaskulitis oder Kollagenose. Auch die Liquordiagnostik ist üblicherweise im spezifischen Fall unergiebig, ist aber – insbesondere im Falle der neurologischen Erstmanifestation einer Vaskulitis oder Kollagenose – zum Ausschluss anderer Differenzialdiagnosen notwendig bzw. ist dann natürlich gemeinsam mit den Ergebnissen supplementärer Laboruntersuchungen diagnostisch richtungsweisend bzw. bestätigend (Tab. 1).
Bildgebende Verfahren
Die zerebrale/spinale MRT (inklusive spezieller Untersuchungsprotokolle wie diffusionsgewichteter Sequenzen oder MR-Angiographie) ist der Goldstandard neuroradiologischer Untersuchungen bei immunologisch bedingten Systemerkrankungen mit neurologischer Manifestation. Insbesondere bei zerebrovaskulären Erkrankungen im Rahmen einer Vaskulitis/Kollagenose ist die Neurosonographie unabdingbarer Bestandteil der diagnostischen Abklärung. Für differenzialdiagnostische Abklärungen, aber auch zur Detektion einer weiteren Organbeteiligung, beispielsweise bei Neurosarkoidose, werden bildgebende Screening-Methoden (Thorax-, Abdomen-CT, FDG-SPECT, Gallium-Szintigraphie) eingesetzt.
Andere diagnostische Maßnahmen
Bei neurologischer (Erst-)Manifestation einer immunologischen Multisystemerkrankung ist gelegentlich zum Ausschluss anderer Differenzialdiagnosen und unter Umständen zur schlussendlichen Diagnosesicherung eine Biopsie (Gehirn, Meningen, Myelon, Nerv/Muskel, Temporalarterie) notwendig. Manchmal ist auch die Biopsie einer anderen Organmanifestation (z. B. Lymphknoten, Haut) zielführend, weil logistisch einfacher und weniger risikobehaftet.
Wesentlich ist die interdisziplinäre diagnostische Abklärung, nicht nur bei manifesten nichtneurologischen Symptomen, sondern auch bei berechtigter Antizipation solcher im weiteren Verlauf. Die interdisziplinäre Abklärung dient nicht nur der Bestätigung bzw. dem Ausschluss anderer Systembeteiligungen, sondern ist auch die Saat für das interdisziplinäre und gemeinsame weitere Vorgehen in Betreuung, Therapie und Monitoring der PatientInnen.
Neuropsychiatrischer systemischer Lupus erythematodes
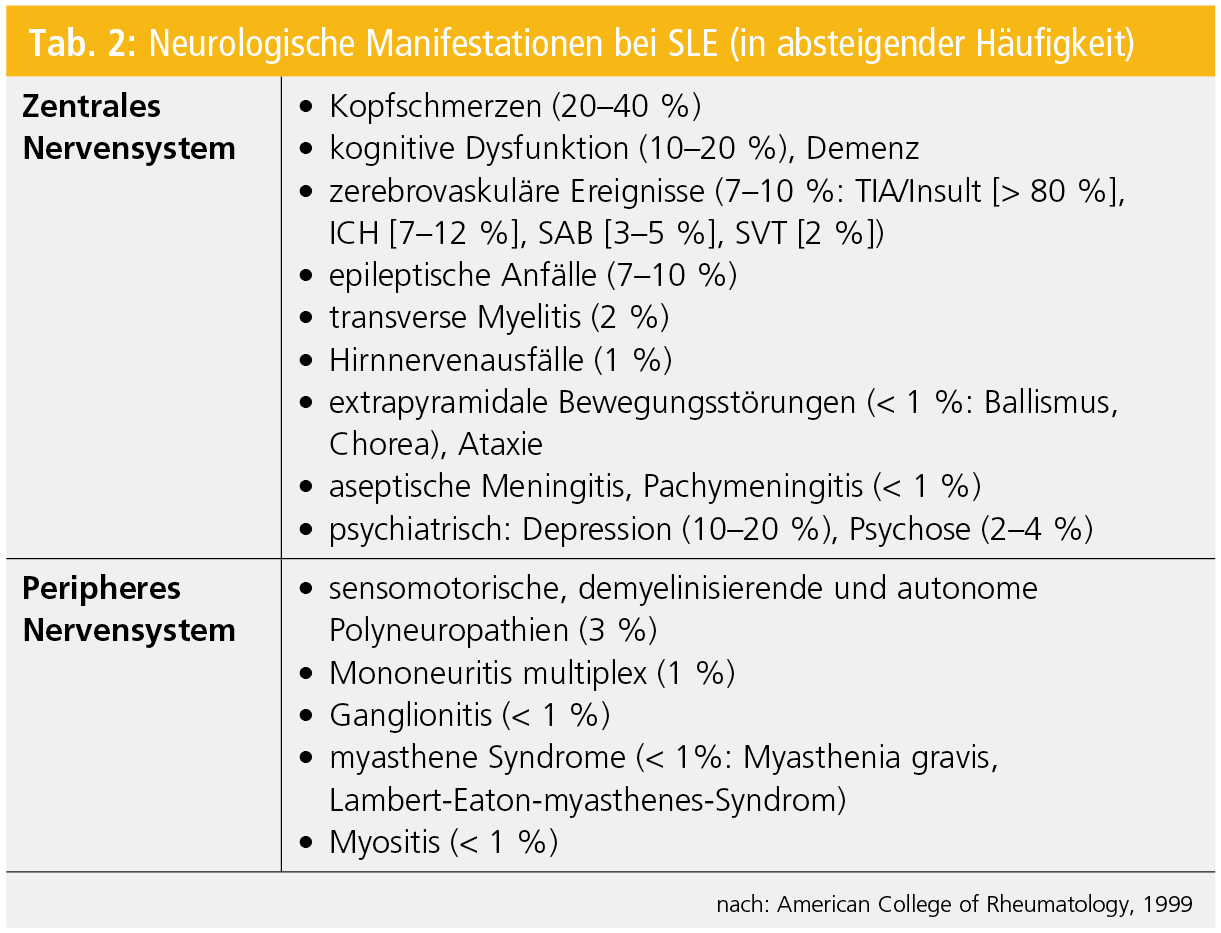
Der systemische Lupus erythematodes (SLE) ist nicht nur wegen seiner Prävalenz (130/100.000), sondern auch aufgrund seiner Symptomenvielfalt die (auto-)immune Multisystemerkrankung. Im Rahmen dieser chronischen Erkrankung mit variablem klinischem Schweregrad können praktisch alle Organe betroffen sein. Neurologische Manifestationen treten initial bereits bei 5 % der SLE-PatientInnen auf und innerhalb der ersten 5 Jahre nach SLE-Diagnose haben 40–60 % der PatientInnen neurologische Symptome (Tab. 2; Abb. 2).
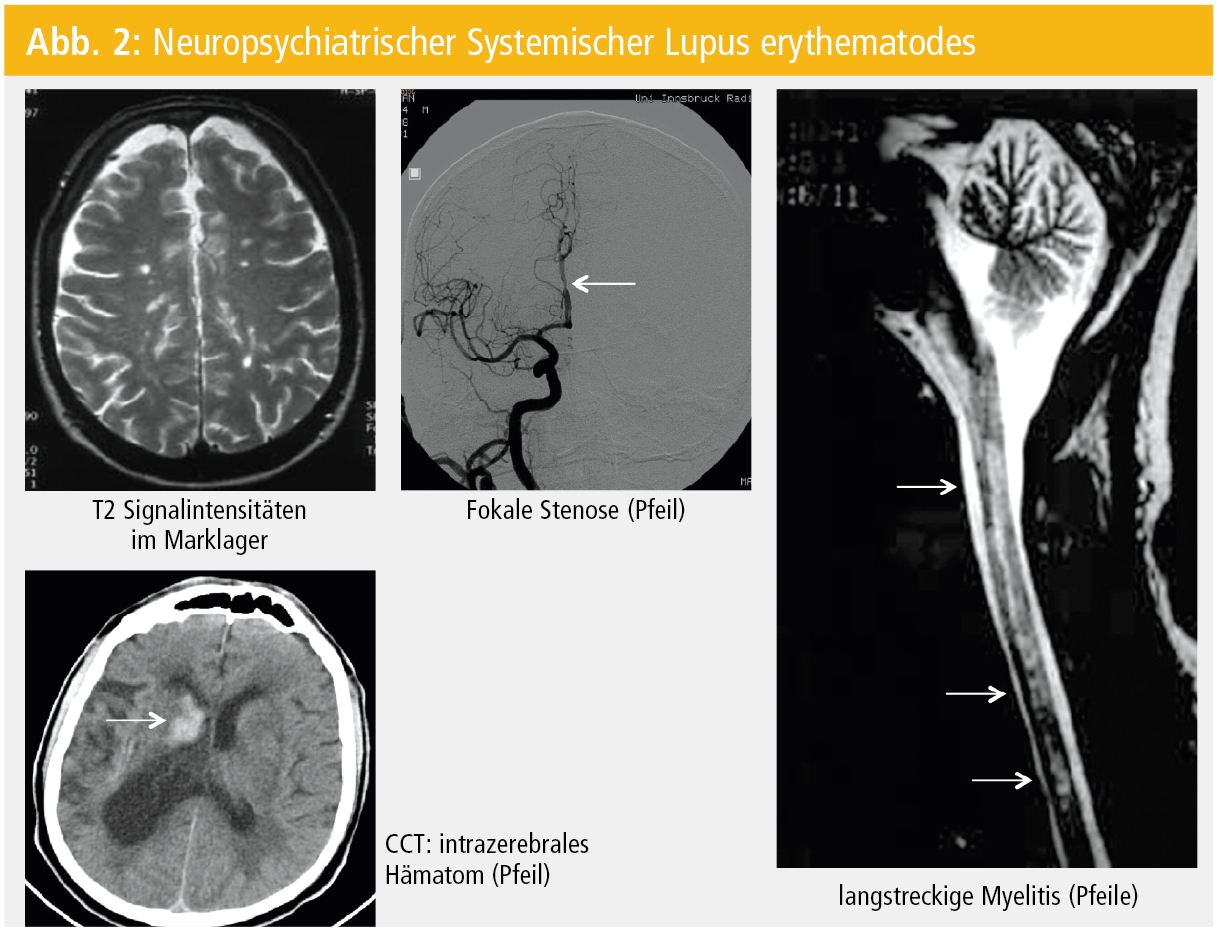
Die Ursache dieser Autoimmunerkrankung ist noch nicht geklärt. Zur Ätiologie des neuropsychiatrischen SLE (NPSLE) haben sich aufgrund heterogener neuropathologischer Befunde im ZNS (überwiegend vaskulopathische Veränderungen an kleinen Gefäßen und diffuse parenchymatöse Läsionen) und PNS (überwiegend Vaskulitis mit inflammatorischen, auch nekrotisierenden Infiltraten, Intimaproliferationen und luminalen Okklusionen) zwei pathogenetische Konzepte, die sich gegenseitig bedingen bzw. ergänzen, entwickelt:
- Vaskulopathisches Konzept:
- Mikro-, seltener Makroangiopathie mit lokaler Ischämie (Thromben, Embolien – bei Libman-Sacks- Endokarditis) mit nachfolgender axonaler Degeneration und sekundärer Demyelinisierung
- vaskulopathische Komplikationen sekundär zu: systemischen SLE- Manifestationen (hämorrhagische Diathese, renale Hypertension, hypertensive Enzephalopathie); opportunistischen Infektionen; medikamentös-toxisch
- Immunpathogenetisches Konzept:
- Intrathekale Produktion von Zytokinen (IL-2, IL-6, IL-10), Chemokinen und Matrixmetalloproteinasen
- Aktivierung von Endothelzellen (Expression von Adhäsionsmole- külen und MHC II)
- humorale Immunreaktionen: Antiphospholipid-Antikörper, Antikörper gegen Endothelzellen, Glutamatrezeptoren und Ribosomen, aber auch gegen (unzählige) andere Antigene des ZNS und PNS; Ausbildung von Immunkomplexen und dadurch mediierte Vaskulitis; Komplementaktivierung
- Nachweis von CD4+-T-Zellen; T-Zell-mediierte Vaskulitis?
Das Auftreten von bzw. die Assoziation mit neuropsychiatrischen Symptomen bei SLE ist von drei Risikofaktoren dominiert:
- Der Schweregrad des SLE per se, definiert durch das Ausmaß an Organmanifestationen und Krankheitsaktivität. PatientInnen mit SLE haben gegenüber der Normalbevölkerung ein ca. 8-fach höheres Risiko für einen ischämischen Insult, insbesondere auch im jüngeren Erwachsenenalter.
- Frühere oder bestehende neuropsychiatrische Symptome im Rahmen des SLE, insbesondere zerebrovaskuläre Ereignisse und epileptische Anfälle betreffend.
- Das Vorhandensein von Antiphospholipid-Antikörpern, die mit einem erhöhten Risiko für zerebrovaskuläre Erkrankungen, epileptische Anfälle und Myelitis assoziiert sind.
Diagnostik
Die diagnostische Abklärung neuropsychiatrischer Symptome bei suspizierten NPSLE erfolgt primär nicht anders als üblich bei solchen Symptomen anderer Genese. In Anhängigkeit des präsenten neurologischen Beschwerdebildes kommen folgende diagnostische Verfahren zum Tragen:
- die MRT (zerebral, spinal) ist der bildgebende Goldstandard mit folgendem Untersuchungsprotokoll: konventionelle T1-/T2-, FLAIR-, diffusions- und kontrastmittelverstärkte T1-gewichtete Sequenzen. Am häufigsten werden dann kleine, fokal subkortikal (zu 80 % frontoparietal) und periventrikulär lokalisierte T2-Signalhyperintensitäten detektiert.
- Liquordiagnostik (Tab. 1): entweder normaler Liquorbefund oder mildes, unspezifisches entzündliches Liquorsyndrom (milde Pleozytose, geringe Erhöhung des Gesamteiweißes, manchmal oligoklonale Banden)
- Laboruntersuchungen: komplettes Blutbild, Differenzialblutbild, Gerinnungsstatus, CRP, BSG, Leber- und Nierenfunktionsparameter, Antiphospholipid-Antikörper, ANA (mit Subsets) …
- Elektrophysiologische Untersuchungen (EEG, NLG, EMG)
- Neuropsychodiagnostik
- Neurosonographie
- Psychiatrische, dermatologische, augenärztliche, kardiologische … Untersuchungen
Therapie
Die kausale Therapie des NPSLE richtet sich nach der relevanten Frage, ob die neurologische Symptomatik nun eher vaskulopathisch/thrombotisch oder inflammatorisch verursacht ist. Eine einfache Faustregel ist bei dieser Entscheidung hilfreich: Fokale neurologische Prozesse sind üblicherweise thrombotisch (insbesondere auch bei Nachweis von Antiphospholipid-Antikörpern) verursacht, während hingegen diffuse Symptome durch Entzündungsmediatoren bzw. Antikörper bedingt sind. Zusätzlich müssen alle konkomitanten Risikofaktoren (z. B. arterielle Hypertonie, Infektionen) gleichzeitig adäquat behandelt werden.
Die Therapie des NPSLE gliedert sich in 4 Bereiche und ist umso effektiver, je interdisziplinärer die Betreuung und Behandlung des betroffenen Patienten/der betroffenen Patientin ist (Tab. 3). Grundsätzlich existieren zur spezifischen Therapie des NPSLE nur wenige kleine kontrollierte Studien. Wenn eine inflammatorische Aktivität im Vordergrund steht, dann erfolgt im Allgemeinen eine primäre Therapie mit Kortikosteroiden, alleine oder in Kombination mit einem Immunsuppressivum (meist Azathioprin oder Zyklophosphamid). Bei therapierefraktären neuropsychiatrischen Manifestationen wird die Therapie eskaliert (Plasmapherese, Rituximab, andere Immunsuppressiva). Wenn zerebrovaskuläre Ereignisse, vor allem TIA/Insult, in Assoziation mit positiven Antiphospholipid-Antikörpern aufgetreten sind, dann wird mit Thrombozytenfunktionshemmern und/oder Antikoagulation (Warfarin, INR 2.0–3.0) behandelt.

Arteriitis temporalis Horton
Diese Riesenzellarteriitis (RZA) ist eine chronische Vaskulitis der großen und mittleren Gefäße. Nachdem diese RZA nicht nur die Arteria temporalis und angrenzende Gefäße, sondern auch beispielsweise die Aorta betrifft und in bis zu 50 % der Fälle mit einer Polymyalgia rheumatica assoziiert ist, gilt die RZA als Systemerkrankung. Mit einer Inzidenz zwischen 1–30/100.000 (je nach geografischer Lage) ist die RZA die häufigste Vaskulitis bei Personen jenseits des Alters von 50.
Die Ätiologie dieser Vaskulitis ist unbekannt, es werden aber auf Basis einer genetischen Prädisposition (HLA-DRB1*04) exogene Trigger, insbesondere Infektionen (Mykoplasmen, Chlamydien, Parvovirus 19) hypothetisiert. Histopathologisch finden sich in 60–70 % der Gefäßbiopsate in allen 3 Gefäßwandschichten inflammatorische Infiltrate (T-Zellen, Makrophagen, dendritische Zellen), die dann die typischen Riesenzellgranulome in der Media ausbilden. Durch Intimahyperplasie wird das arterielle Gefäßlumen partiell oder komplett okkludiert (Abb. 3), wodurch es zu den ischämischen Manifestationen der RZA kommt.
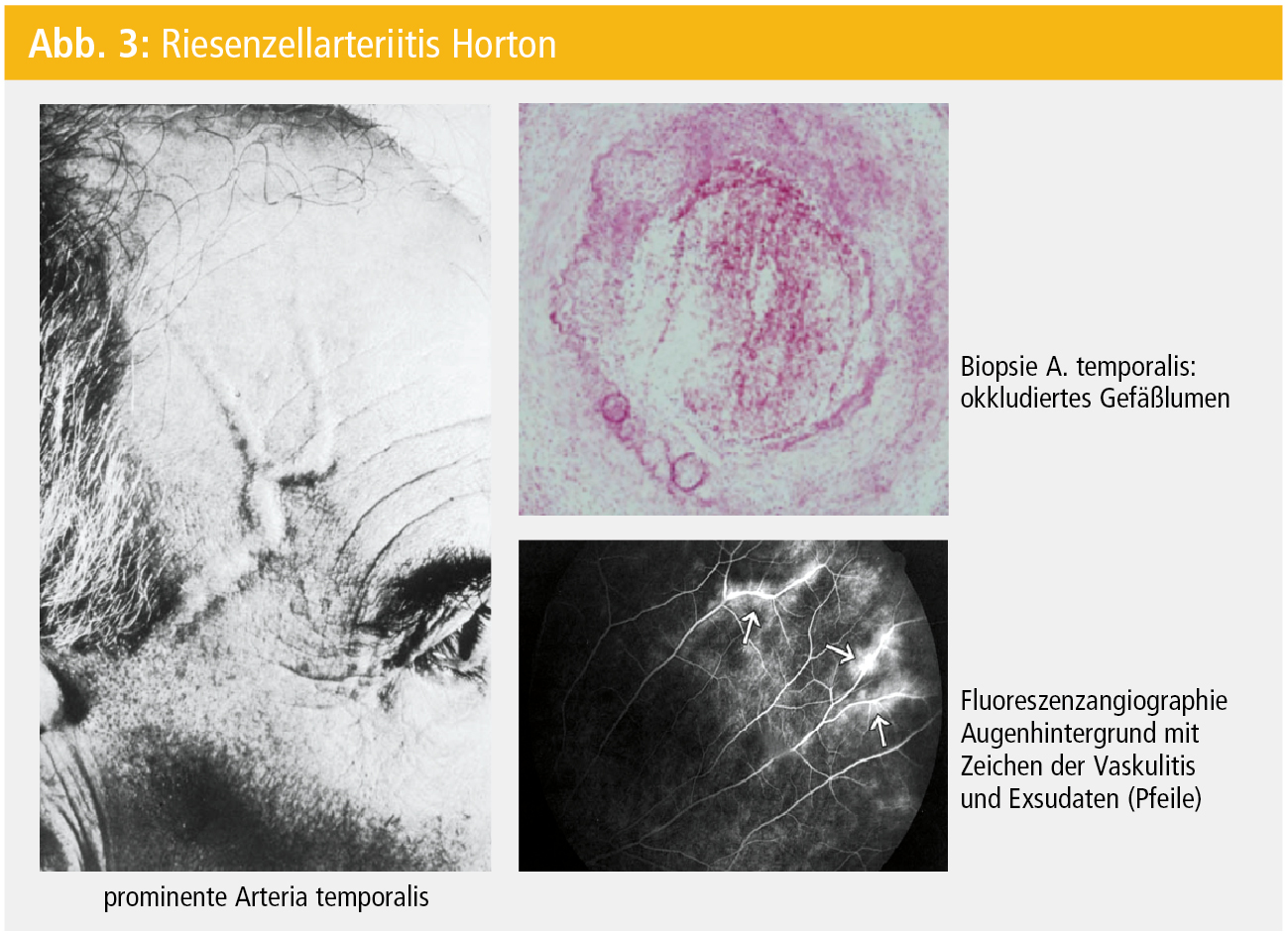
Klinisch (Tab. 4) präsentiert sich die RZA Horton am häufigsten als typische Arteriitis temporalis (Abb. 3), oft verbunden mit Kopfschmerzen und einer Claudicatio masticatoria. Das unmittelbare Risiko besteht in einer ischämischen Komplikation mit Visusverlust (Abb. 3). Schon ein Drittel der PatientInnen hat zeitgleich mit der beginnenden Arteriitis temporalis eine Visusstörung, zumeist eine Amaurosis fugax. Diese wiederum ist ein absolutes Warnsignal und Vorbote für einen drohenden permanenten (und dann irreversiblen) Visusverlust, aber auch für (wesentlich seltenere) zerebrovaskuläre Ereignisse (TIA/Insult).

Diagnostik
Die diagnostische Abklärung hat unmittelbar bei Verdacht zu erfolgen. Die gängige Praxis fordert die diagnostische Evidenz durch die Biopsie der Temporalarterie vor Beginn der Kortikosteroidtherapie. In Abhängigkeit der initialen klinischen Symptomatik und insbesondere des Risikos einer drohenden Komplikation (z. B. Visusverlust) ist aber das Zuwarten auf den Termin bzw. das Ergebnis der Biopsie ohne Beginn der Kortikosteroidtherapie nicht zulässig, weil potenziell den Patienten/die Patientin gefährdend. Außerdem haben Studien gezeigt, dass die Steroidgabe bereits ab einer Woche vor der Biopsie keine wesentliche Auswirkung auf den histopathologischen Befund hat. Und schließlich ist die Ultraschalldiagnostik der A. temporalis (und angrenzender Arterien) bereits so ausgereift, dass mit einer Spezifität von ca. 90 % und einer Sensitivität von > 75 % das sogenannte „Halo-Zeichen“ (echoarme, zirkumferente Verdickung der luminalen Gefäßwand) diagnostisch ausschlaggebend ist. In Anbetracht der Häufigkeit (und Gefahr) einer Augenbeteiligung ist eine ophthalmologische Untersuchung obligatorisch.
Therapie
Der Therapiebeginn mit Kortikosteroiden sollte so früh wie möglich sein, um drohende Komplikationen zu verhindern: initial 40–60 mg/Tag oder 1 mg/kg Körpergewicht/Tag, nach 2–4 Wochen (innerhalb dieser Zeit remittieren üblicherweise die ursprünglichen Symptome) dann Reduktion der Kortikosteroiddosis auf die niedrigstmögliche Erhaltungsdosis für 1 bis 2 Jahre. Bei 40–50 % der PatientInnen kommt es während (oder trotz) Steroiderhaltungsdosis zu einem neuerlichen Krankheitsschub. In dieser Situation, aber auch als steroidsparende Rationale, kommen dann immunsuppressive Therapien (mit bislang aber bescheidener Studienevidenz) zum Einsatz: Azathioprin, Methotrexat, TNF-alpha-Inhibitoren (Etanercept) und monoklonale Antikörper (Tocilizumab, Anti-IL-6R). Adjuvant wird auch eine niedrig dosierte Aspirin-Gabe empfohlen, weil sich in einer retrospektiven Analyse gezeigt hat, dass dadurch ischämischen Komplikationen erfolgreich vorgebeugt werden konnte.
Neurosarkoidose
Die Sarkoidose ist eine histopathologisch und klinisch charakteristische granulomatöse Multisystemerkrankung unklarer Ätiologie, die mit einer Prävalenz von 50/100.000 grundsätzlich jedes Organ betreffen kann, sich jedoch mehrheitlich (90 %) pulmonal manifestiert.
Klinisch verläuft die Neurosarkoidose (bis zu 15 % der Fälle) in Schüben mit Remissionen im Intervall (und imitiert somit a priori differenzialdiagnostisch eine Multiple Sklerose) oder auch chronisch progredient.
Die neurologische Symptomatik hängt von der Topographie der Granulome ab, am häufigsten zeigt sich eine basale granulomatöse Meningitis mit Hirnnervenbeteiligung (70 %), wobei vornehmlich der Nervus facialis (auch als Sonderform Heerfordt-Waldenström-Syndrom mit Fieber, Parotitis, Uveitis anterior und Fazialisparese) bzw. der Nervus opticus (oft auch beidseitig) betroffen sind. Neben der aseptischen Meningitis (und der äußerst seltenen damit verbundenen akuten Komplikation eines Hydrozephalus) kann die Sarkoidose auch Multiple Sklerose-ähnliche Läsionen im periventrikulären Marklager imitieren, selten auch als tumefaktive Läsionen mit lokal raumfordernder Wirkung imponieren.
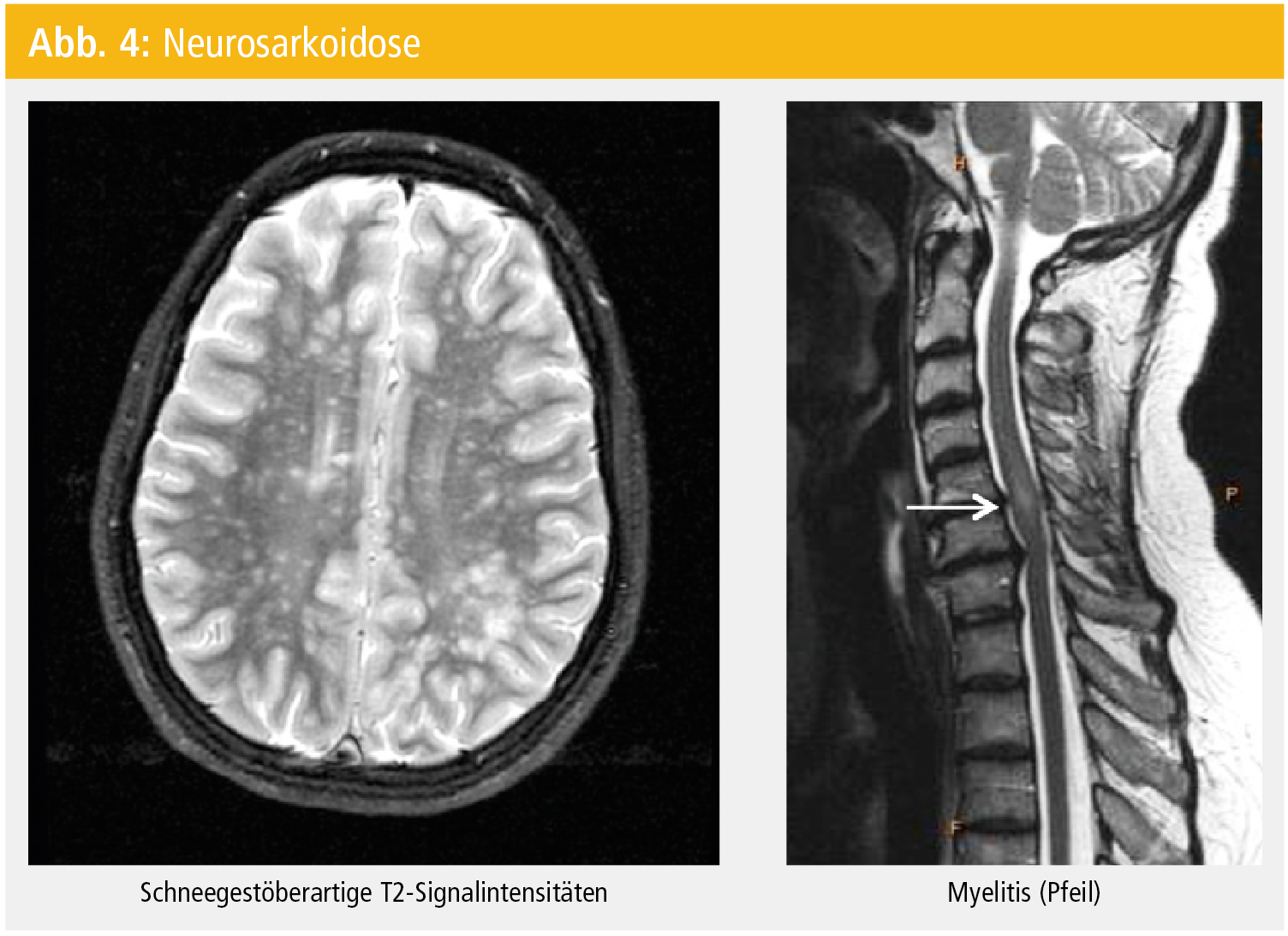
Die hingegen diffuse Infiltration des Hirnparenchyms verursacht eine Leukenzephalopathie. Selten manifestieren sich Granulome in der Hypophyse mit entsprechend endokriner Dysfunktion (Diabetes insipidus, Amenorrhö). In bis zu einem Drittel der Fälle kommt es auch zur spinalen Manifestation (Abb. 4), die dann klinisch einer subakuten transversen Myelitis entspricht und in diesem Falle als Erstmanifestation einer Sarkoidose die differenzialdiagnostische Abgrenzung gegenüber Raumforderungen oder auch entzündlich-demyelinisierenden Erkrankungen (spinale Multiple Sklerose oder – bei langstreckigen Myelonläsionen – Neuromyelitis optica) erschwert.
Diagnostik und Therapie
Bei bekannter Sarkoidose und nunmehr neurologischer Symptomatik ist (abgesehen vom Ausschluss anderer Differenzialdiagnosen, wie z. B. opportunistischer Infektion im Rahmen einer immunsuppressiven Therapie der Sarkoidose) in erster Linie die zerebrale/spinale MRT-Untersuchung diagnostisch relevant (Abb. 4).
Die größere diagnostische Herausforderung ist aber der Verdacht einer Neurosarkoidose ohne bekannte systemische Manifestation (Tab. 5). Hier sind ebenfalls in erster Linie bildgebende Verfahren einzusetzen: Thorax-CT zur Detektion einer pulmonalen oder Lymphknotenbeteiligung, gegebenenfalls FDG-PET zur Identifizierung von sarkoiden Granulomen in anderen Organen und natürlich erneut die MRT. Signalintensitäten, mitunter kontrastmittelspeichernd, können sich leptomeningeal, supra-/infratentoriell, spinal und entlang von Mittellinienstrukturen (Hypophyse, Hypothalamus) finden.
Als Caveat gilt aber, dass ein unauffälliger MRT-Befund eine Neurosarkoidose nicht ausschließt, insbesondere bei reinem Hirnnervenbefall oder unter laufender Steroidtherapie (und da wird es dann auch noch gegenüber der differenzialdiagnostischen Überlegung eines ZNS-Lymphoms zusätzlich schwierig).
Die Liquordiagnostik ist unspezifisch und die mancherorts propagierte Bestimmung des ACE (Angiotensin Converting Enzymes) im Liquor ist völlig unzuverlässig und daher zur Diagnose oder zum Ausschluss einer Neurosarkoidose nicht geeignet. Aufgrund der unspezifischen neuroradiologischen und Liquorbefunde ist bei einer isolierten Neurosarkoidose ohne jeglichen Nachweis einer systemischen Beteiligung die diagnostische Sicherheit durch eine Biopsie (eines parenchymatösen Granuloms oder der Meningen) prinzipiell anzustreben, aber häufig aufgrund der Unzugänglichkeit der Läsion oder bei mangelnder Erfolgsaussicht (z. B. bei meningealer Biopsie) frustran.
Therapie
Die Therapie der Neurosarkoidose folgt (in Ermangelung kontrollierter Studien) im Wesentlichen der üblichen Vorgehensweise bei immunologischen Erkrankungen: Gabe von Steroiden als Basistherapie und Immunsuppressiva als Therapieeskalation (Tab. 6).

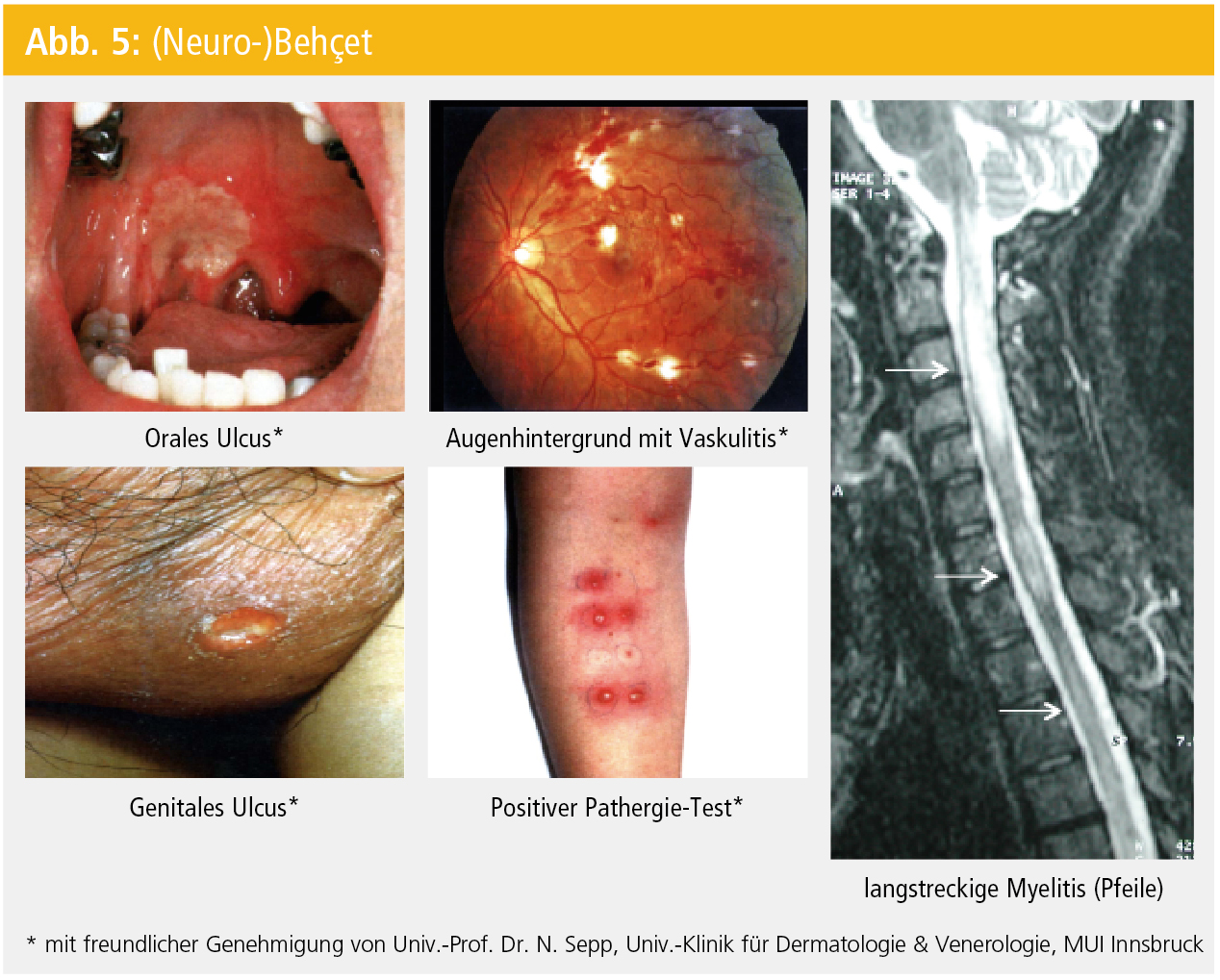
Neuro-Behçet
Diese Systemerkrankung ist nach der Erstbeschreibung durch den türkischen Arzt Hulusi Behçet (1889–1948) benannt („Über rezidivierende, aphthöse, durch ein Virus verursachte Geschwüre am Mund, am Auge und an den Genitalien“, Deutsche Dermatologische Wochenschrift, 1937).
Die Prävalenz in Ländern entlang der Seidenstraße, vor allem der Türkei, beträgt 80–400/100.000, in Mitteleuropa hingegen nur 0,5/100.000.
Die Ätiologie dieser entzündlichen Systemerkrankung (Vaskulitis) ist unklar, es wird auf Basis einer genetischen Suszeptibilität (HLA-B51) eine durch einen infektiösen Trigger ausgelöste Autoimmunreaktion gemutmaßt.
Die klinischen Symptome (Tab. 7) sind – der Erstbeschreibung entsprechend – dermatologische (orale und genitale Aphthen bzw. Ulzera; akneiforme oder Erythema nodosum-ähnliche Effloreszenzen) und ophthalmologische (anteriore oder posteriore Uveitis, retinale Vaskulitis) Manifestationen (Abb. 5). Neurologische Symptome können als Erstmanifestation eines Mb. Behçet (z. B. durch Myelitis; Abb. 5), im Rahmen der systemischen Organbeteiligung (z. B. durch zerebrovaskuläre Manifestationen wie Sinusvenenthrombose, zerebrale Ischämien, intrakranielle Blutungen) oder als sekundäre Folgen (z. B. toxische Nebenwirkungen bei immunsuppressiver Behandlung des Mb. Behçet) auftreten.
Diagnostik und Therapie
Abgesehen von der Labor- und Liquordiagnostik (in erster Linie zu differenzialdiagnostischen Zwecken) bzw. MRT-Verfahren ist der positive Pathergie-Test (Abb. 5) für die Diagnose eines Mb. Behçet richtungweisend (und somit auch Teil der diagnostischen Kriterien; Tab. 8).

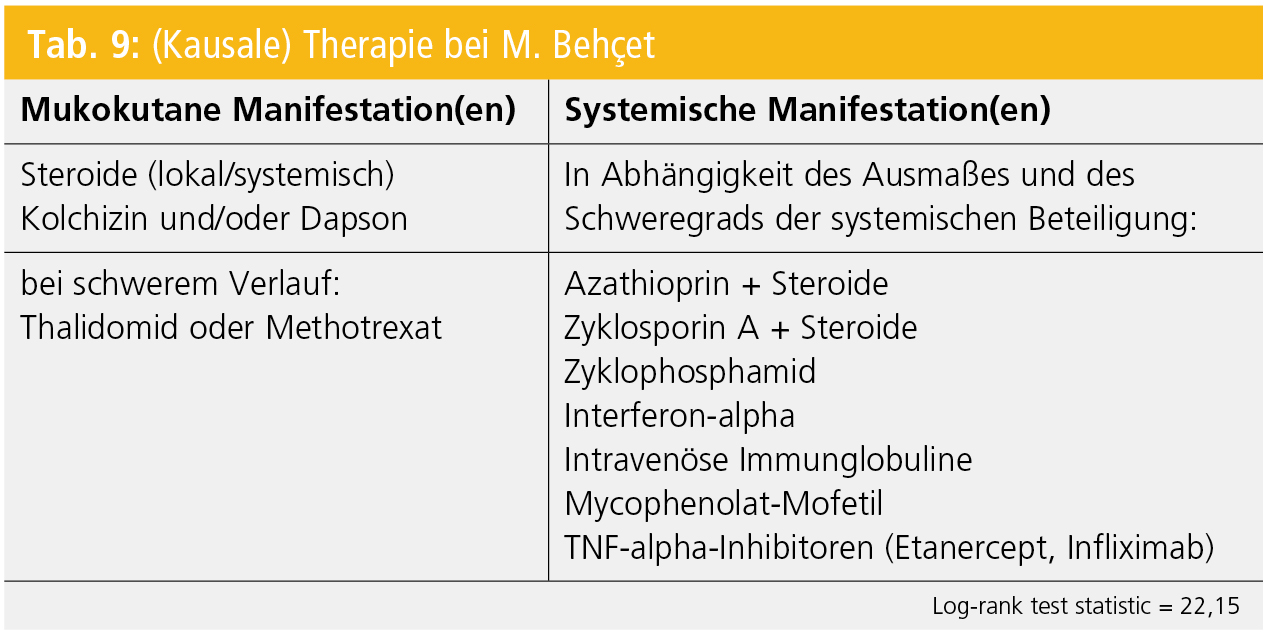
Die Therapie richtet sich nach dem Schwergrad, d. h. nach dem Ausmaß der systemischen Beteiligung, und vereint kausale (immunsuppressive, -modulierende) und symptomatische Therapien (Tab. 9).
Weiterführende Literatur:
– Aringer M et al., Current state of evidence on „off-label“ therapeutic options for systemic lupus erythematosus, including biological immunosuppresive agents, in Germany, Austria and Switzerland – a consenus report. Lupus 2012; 21:386–401.
– Bertsias GK, Boumpas DT, Pathogenesis, diagnosis and management of neuropsychiatric SLE manifestations. Nature Rev Rheumatol 2010; 6:358–367.
– Bertsias GK et al., EULAR recommendations for the management of systemic lupus erythematosus with neuropsychiatric manifestations: report of a task force of the EULAR standing committee for clinical affairs. Ann Rheum Dis 2010; 69:2074–2082.
– Borchers AT, Gershwin ME, Giant cell arteritis: a review of classification, pathophysiology, geoepidemiology and treatment. Autoimmunity Rev 2012; 11:A544–A554.
– Chen M, Kallenberg CGM, ANCA-associated vasculitides – advances in pathogenesis and treatment. Nature Rev Rheumatol 2010; 6:653–664.
– Holzapfel R, Mäurer M, Neurosarkoidose. J Neurol Neurochir Psych 2011; 12:280–283.
Spezielle Literatur beim Verfasser

AutorIn: Univ.-Prof. Dr.
Thomas Berger, MSc
Leiter der AG Neuroimmunologie und Multiple Sklerose, Universitätsklinik für Neurologie, Medizinische Universität Innsbruck

Ursprünglich erschienen:
neuro 04|2012
neuro 04|2012

