





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Update zur Diagnostik von Demenzen
14. Dezember 2012
Rund 36 Millionen Menschen weltweit und etwa 130.000 in Österreich leiden an Demenz, gekennzeichnet durch fortschreitende Abnahme von Hirnleistungen in mehreren kognitiven und anderen Bereichen, häufig begleitet von Verhaltensstörungen und neuropsychiatrischen Symptomen. Weltweit wird ihre Zahl bis 2050 auf etwa 100 Millionen ansteigen (46 % Asien, 30 % Europa, 12 % USA) mit stärkster Zunahme in Entwicklungsländern (70 %). Für Österreich wird ein Anstieg auf etwa 250.000 angenommen.
Die Alzheimer-Kankheit (AD) stellt die häufigste Demenzform (60 %), gefolgt von Demenz mit Lewy-Körpern (DLB, 15–29 %), vaskulärer Demenz (VaD, rund 10 %) und anderen Prozessen (10–15 %), am häufigsten sind Mischformen (50-70%). Sogenannte gutartige Altersvergesslichkeit („mild cognitive impairment“, MCI) als früheste Manifestation kognitiver Störungen mit nur geringen neuropsychologischen Defiziten als Vorstadium der AD und anderer Demenzen zeigt eine mittlere Prävalenz von 20–30 % mit Demenzentwicklung in 90 % binnen 9–10 Jahren (jährlich 10–15 %).
Klinische Diagnose
Für die klinische Erfassung der wichtigsten Demenzerkrankungen wurden in jüngster Zeit von internationalen Expertengruppen revidierte Konsensuskriterien erarbeitet, die sich auf eine Kombination klinisch-neuropsychologischer Daten, moderner bildgebender Verfahren (strukturelle und funktionelle MRT, multimodale Kombination von MRI und PET sowie Nachweis von β-Amyloidablagerungen im Gehirn [PIB-PET]) und von Biomarkern (im Liquor cerebrospinalis und Plasma) stützen und eine exaktere Diagnose gestatten:
Präklinische Demenz („minimal cognitive impairment“)
Für die Diagnose der präklinischen AD wurde von einer Arbeitsgruppe des National Institute on Aging und der Alzheimer-Society (NIA-AA) unter Verwendung moderner bildgebender Verfahren (FDG-PET, Amyloidnachweis) und Biomarker mehrere Stadien vorgeschlagen (Tab. 1), nach denen 97 % kognitiv intakte Personen klassifiziert wurden und nur 3 % unklassifiziert blieben.

Alzheimer-Krankheit
Für die Diagnostik der AD wurden neben den revidierten NINCDS-ADRDA-Kriterien kürzlich revidierte EFNS-Kriterien vorgelegt, die sich auf klinische Untersuchungsbefunde, Erfassung kognitiver Funktionen durch neuropsychologische Testung, Aktivitäten des täglichen Lebens, Komorbiditäten, Blutbefunde, bildgebende Verfahren (MRT – Nachweis von Hippokampusatrophie, PET – Amyloidablagerungen), Abnahme von Sauerstoffverbrauch und Glukosestoffwechsel sowie Liquoranalyse (Anstieg von Tau-Protein; Abnahme von β-Amyloid) stützen . Betont sei, dass eine Abnahme von Aβ im Liquor bereits 5–10 Jahre vor dem Beginn der Demenz nachweisbar ist. Daneben tragen Proteinbiomarker im Blut zur Diagnose von MCI und AD bei. Hinsichtlich der aktuellen Definition der AD siehe Tabelle 2.

Die kürzlich vorgeschlagene Revision diagnostischer Kriterien für die AD zeigt die Tabelle 3. Die aktuellen Fakten zur AD wurden kürzlich durch die US Alzheimer Association zusammengefasst, wie auch die Bedeutung der Biomarker für deren Diagnostik und Therapie.

Vaskuläre Demenz
Das diagnostische Vorgehen bei vaskulär bedingten kognitiven Störungen (vaskuläre Demenzen) umfasst den Nachweis eines Demenzsyndroms bzw. von MCI, den Nachweis einer zerebrovaskulären Erkrankung mittels bildgebender Verfahren und den Nachweis für den Zusammenhang zwischen beiden.
Man unterscheidet verschiedene Subtypen:
- Post-Stroke-Demenz mit plötzlichem Beginn fokaler neurologischer Symptome und kognitiver Beeinträchtigung, die sich rückbilden kann. Prävalenz etwa 30 %, doch zeigen 7–16 % bereits präexistente Kognitionsstörungen;
- Multiinfarktdemenz mit langsamer Entwicklung nach oft stummen Infarkten, langsamen Plateaus zwischen den Ereignissen und Fluktuationen;
- Strategische Infarktdemenz mit kleinen Infarkten in strategisch wichtigen Hirnarealen (Thalamus, Stammkerne, Marklager); kognitive Defizite gehen mit Apathie und Verhaltensstörungen einher;
- Subkortikale vaskuläre Demenz als häufigste Form durch Erkrankung kleiner Hirngefäße mit Lakunen und Marklagerschäden zeigt eine oft über Jahre verlaufende langsame Demenzentwicklung mit exekutiven Dysfunktionen, weniger starken Gedächtnisdefiziten sowie neurologischen Zeichen, Gang- und Miktionsstörungen sowie psychomotorischer Verlangsamung.
Die klinischen Kriterien für die Diagnose der VaD zeigen eine durchschnittliche Sensitivität von 0,49 und Spezifität von 0,86 auf. Die Rolle von PET-Untersuchungen in der Differenzialdiagnose der VaD wurde kürzlich zusammengefasst. Unter Ergänzung bisheriger diagnostischer Kriterien (ICD-10; SCADDTD, NINDS-AIREN) wurde für die klinische Diagnose vaskulärer Demenzen eine Definition vorgeschlagen, die weitere Standardisierung und Validierung erfordert (Tab. 4).

Demenz mit Lewy-Körpern und Parkinson-Demenz
Die klinische Diagnose der DLB als der zweithäufigsten Ursache neurodegenerativer Demenzen umfasst eine Kombination progressiver kognitiver Störungen vorwiegend von visuospatialen Fähigkeiten und frontal-exekutiven Funktionen, visuellen Halluzinationen und deutlichen Fluktuationen von Wachheit und Aufmerksamkeit mit häufiger Neuroleptikaintoleranz, ferner Parkinson-Symptome vorwiegend vom rigid-akinetischen Typ mit nur seltenem Ruhetremor. Häufig bestehen frühe Zeichen von Verhaltensstörungen im REM-Schlaf („rapid eye movement“) wie Schreien, Sprechen und motorische Übererregung).
DLB ist von Parkinson-Demenz (PDD) abzugrenzen, wobei sich dies nach aktuellem Konsens auf den Beginn der Demenz gegenüber motorischem Parkinson ( 1 Jahr PDD) gründet. Beide Syndrome werden als verschiedene Phänotypen eines Krankheitsspektrums mit nur geringen klinischen und morphologischen Unterschieden interpretiert. Wesentliche Unterschiede zur AD, vorwiegend in Frühstadien, sind seltener schwere kognitive Störungen (Gedächtnis, verbale, nichtverbale und visuelle Bereiche), stärkere exekutive Dysfunktionen, Aufmerksamkeitsdefizite, visuospatiale Störungen und Sprachdefizite, rascherer klinischer Verlauf und höhere Mortalität.
Differenzialdiagnostisch zeigt die DLB verminderte dopaminerge nigrostriäre Aktivität (SPECT oder PET), geringere Atrophie (MRT) und stärkeren okzipitalen Hypometabolismus gegenüber der AD sowie stärkere frontale Atrophie gegenüber PDD, während α-Synuklein, phosphoryliertes Tau-Protein sowie MHPG (3-Methoxy-4-Hydroxyphenyl-Glykol) im Liquor sowie erhöhter Amyloidgehalt im Kortex für DLB, erhöhter Tau- und verminderter Amyloidgehalt im Liquor für AD sprechen.
Mischtyp-Demenzen
Die Diagnose gemischter Demenzformen wird bei der häufigen Kombination von AD mit zerebrovaskulären Prozessen oder Lewy-Körper-Syndromen gestellt, die wechelseitige Effekte verursachen, wobei vaskulären Faktoren eine wichtige Rolle zukommt. Neben klinischen Symptomen (progressive Demenz, vaskuläre Prozesse, Parkinson-Syndrome) sind bildgebende Befunde (MRT, PET) sowie vaskuläre Risikofaktoren wichtige diagnostische Hinweise.
Frontotemporale Demenzen
Diese klinisch-genetisch und morphologisch heterogene Gruppe mit Beginn zwischen 45. und 64. Lebensjahr zeigt früh Persönlichkeitsveränderungen, Antriebsmangel und Perseveration, prominente Sprachprobleme bei gegenüber dem M. Alzheimer geringeren Gedächtnisstörungen bei fehlender Krankheitseinsicht.
Sie umfasst 3 klinischen Untergruppen: a) Verhaltensvariante (bvFTD) mit progredienten Verhaltens- und exekutiven Störungen, b) semantische Demenz (SD) mit Verlust von Objektkenntnis oder motorischen Sprachproblemen mit ausgeprägter Atrophie der vorderen Temporallappen sowie c) progressive nichtfluente Aphasie (PNFA) mit expressiven oder motorischen Sprachdefiziten bei vorherrschender perisylvischer Atrophie links. Ihnen entsprechen nach den vorherrschenden Proteinablagerungen 3 molekulare Formen: 1. Mikrotubulus-assoziertes Tau-Protein (FTLD-Tau), 2. TAR-DNA-bindendes Protein-43 (FTLD-TDP) und 3. Fusioniertes Sarkom-Protein (FTLD-FUS). Bis zu 40 % der FTD-PatientInnen haben familiären Hintergrund und 30–50 % der familiären Fälle eine autosomal-dominante Vererbung; daneben wurden verschiedene Genmutationen beschrieben. Die Kombination von Bildgebung (frontale und frontotemporale Atrophie und Hypometabolismus bei FTD, Hippokampusatrophie und parietale Minderdurchblutung bei AD) und Liquormarkern (Korrelation von Tau zu Aβ-42) gestattet eine Abgrenzung beider Krankheiten.
Prionenerkrankungen
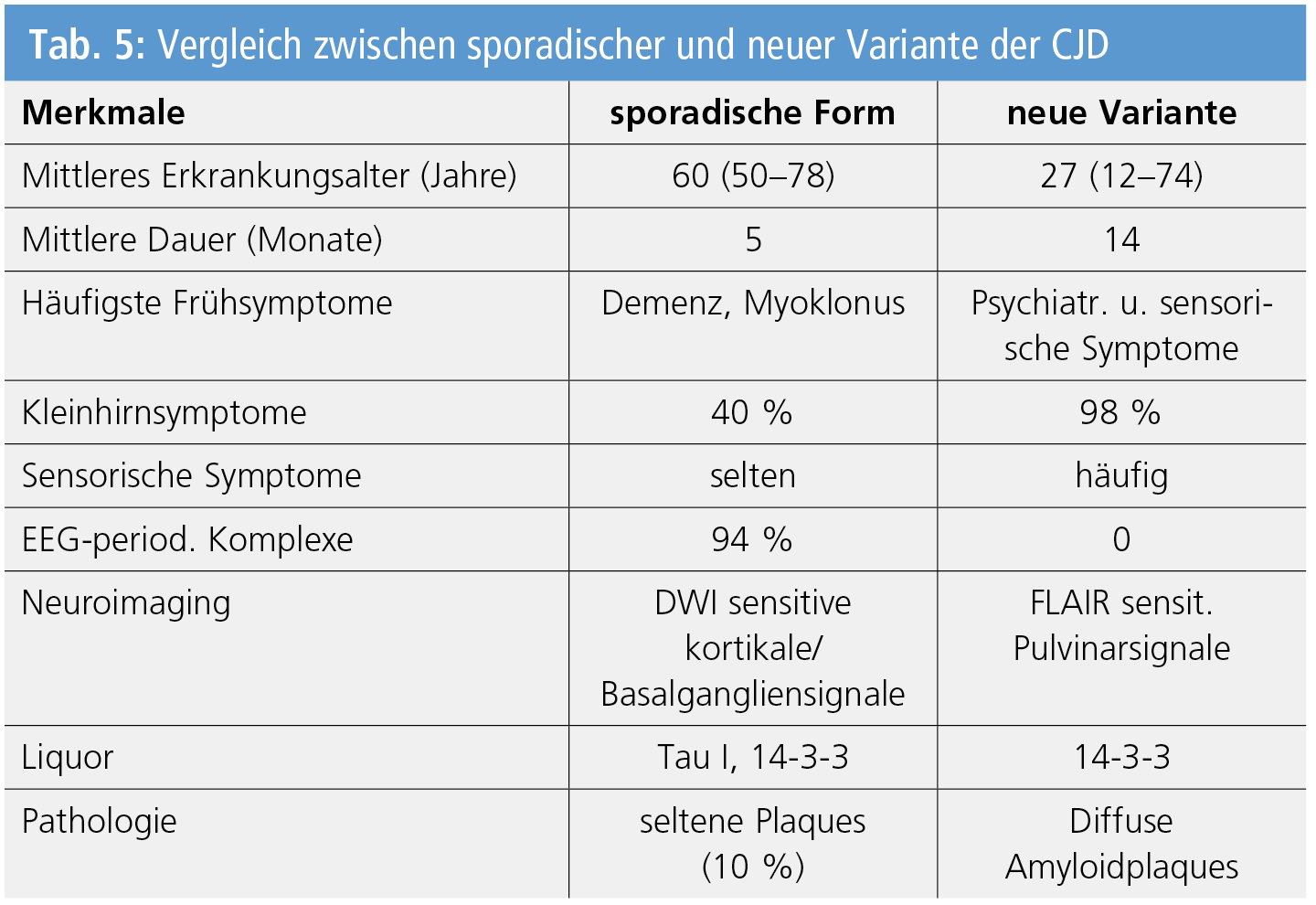
Übertragbare spongiforme Enzephalopathien oder Prionenerkrankungen bei Menschen können sporadisch, familiär oder erworben (durch Infektion) auftreten. Die häufigsten sporadischen Formen umfassen ein breites Spektrum klinisch-pathologischer Varianten mit heterogener Krankheitsdauer, Symptomatologie, Art und regionaler Verteilung der Hirnläsionen. Die Phänotypen sind die sporadische Creutzfeldt-Jakob-Krankheit (CJD) mit Beginn im 50.–78. Lebensjahr und mittlerer Krankheitsdauer von 14 Monaten. Sie zeigt kognitive und Verhaltensstörungen, Gedächtnisverlust, zerebelläre und visuelle Symptomatik, pyramidale und extrapyramidale Symptome, Anfälle, Myoklonien und terminalen akinetischen Mutismus.
Meist bestehen typische EEG-Veränderungen (generalisierte Perioden scharfer oder triphasischer langsamer Wellen), im MRT Signalhyperintensität in Kortex und Striatum sowie signifikant erhöhtes Tau-Protein und 14-3-3-Protein im Liquor. Die neue Variante der CJD (nv-CJD) mit mittlerem Erkrankungsalter von 27 Jahren und mittlerer Dauer von 5 Monaten ohne Hinweise auf familiäre Formen zeigt initiale Verhaltensstörungen, psychiatrische Anomalien, häufig sensorische und zerebelläre Störungen, Fehlen typischer EEG-Veränderungen, bilaterale MRI-Signale im Pulvinar thalami, erhöhtes 13-3-3-Protein im Liquor und häufig positive Tonsillenbiopsien (Tab. 5). Seltenere Formen sind iatrogene CJD (Übertragung durch kontaminierte Instrumente, Duratransplantate, Wachstumshormongaben), sporadische und familiäre Insomnie sowie erbliche Formen (familiäre CJD und Gerstmann-Sträussler-Scheinker-Syndrom). Diesen Phänotypen entsprechen 6 molekular-morphologische Untergruppen, die mit großer Sicherheit zwischen internationalen Überwachungszentren fassbar sind.
Klinisch-pathologische Korrelationen
Die Kombination klinischer und neuropsychologischer Befunde mit modernen bildgebenden Verfahren und aktuellen Biomarkern gestattet die klinische Diagnose der AD mit einer Sicherheit von bis zu 96 %, den Ausschluss nichtdementer Formen in 62–100 % der Fälle bei einer Sensitivität und Spezifität gegenüber anderen Demenzen zwischen 44 und 87 %. Die Kombination der besten Liquor- und MRI-Daten ermöglicht eine bessere klinische Diagnose und wird durch die Anwendung multimodaler Techniken und moderner Biomarker zu einer verbesserten Früherfassung von Demenzprozessen als Voraussetzung für bessere Prävention und Therapie führen. Vor allem der In-vivo-Nachweis von fibrillärem β-Amyloid durch [11C]PIB PET ist wertvoll für die prospektive Erfassung und Voraussage kognitiver Störungen.
Nach wie vor stellt die histopathologische Untersuchung mit Anwendung moderner Immunhistochemie, molekular-biologischer und genetischer Methoden unter Standardbedingungen den „Gold-Standard“ für die Diagnose demenzieller Prozesse, wenngleich sie durch die häufige Überlappung verschiedener Prozesse und die im Alter zunehmende Multimorbidität großen Schwierigkeiten begegnen kann.
Eine kürzlich erstellte Übersichtsarbeit an 2.861 neurodegenerativen Demenzfällen des National Alzheimer’s Coordinating Centers (NACC) ergab eine hohe Diagnosequote für AD (85 % Sensitivität, 51 % Spezifität) bei geringer Sensitivität und Spezifität für DLB. Ein jüngster kritischer Vergleich der Beziehung zwischen pathologischen Alzheimer-Läsionen und kognitivem Status bestätigt erneut deren stärkere Korrelation mit dem Schweregrad kortikaler Neurofibrillendegeneration und geringer mit dem der Amyloidplaques. Letztlich ist der Funktionsverlust und Ausfall von Synapsen durch Schädigung wichtiger neuronaler Netzwerke als wesentliche Demenzursache anzusehen. Neueste Befunde an transgenen Mausmodellen fanden einen Verlust von Dendritendornen noch vor Bildung von Amyloidplaques.
Daneben ist zu betonen, dass sich die Alzheimer-Morphologie bei Senioren über 85 Jahren in Intensität und Verteilung gegenüber Jüngeren stark unterscheidet und häufig Überlappungen zwischen Dementen und Nichtdementen bestehen. Demenz bei den Ältesten korreliert oft nur mäßig mit der Alzheimer-Pathologie, während kardio- und zerebrovaskuläre Komplikationen bei solchen mit nur mäßigen Alzheimer-Läsionen erheblich zur Demenz beitragen und damit oft als Mischtypdemenz imponieren. Ferner entspricht eine große Anzahl dementer Personen nicht den pathologischen Kriterien der AD und ist als „Demenz unklarer Ätiologie“ zu klassifizieren.
Rasch verlaufende Demenzen
Im Vergleich zu den meist chronisch über Jahre verlaufenden typischen Demenzprozessen bieten rasch fortschreitende Demenzen (RPD) mit relativ kurzem, tödlichen Verlauf erhebliche diagnostische Schwierigkeiten. Neben häufigen sporadischen und genetischen Prionenerkrankungen umfassen sie rasch progrediente Tauopathien und Synukleinopathien, Autoimmuninfektionen sowie toxisch-metabolische und Tumorerkrankungen. Sie werden häufig mit CJD verwechselt. Unter über 1.100 Hirnautopsien war AD am häufigsten (50 %), 32 % negativ für Prionenkrankheiten, 12 % vaskuläre Demenzen und 23 % behandelbare Krankheiten; in einer anderen Studie waren 47 % sporadische CJD, 13,6 % erworbene Prionenkrankheiten und 38 % Nichtprionenprozesse. Rasch progredient verlaufende AD kann CJD vortäuschen. Diagnostisch können sensitive Liquormarker oder Hirnbiopsie eine wichtige Rolle spielen, wenngleich Biopsiebefunde bei Demenzen oft unspezifisch sind.
Morphologische Diagnosekriterien
Kürzlich modifizierte Konsensuskriterien für die neuropathologische Diagnose degenerativer Demenzprozesse von US- und europäischen Expertengruppen umfassen:
- morphologische Alzheimer-Läsionen, die bei gesichertem Fehlen kognitiver Störungen vorliegen können,
- den Vorschlag eines gegenüber den bisher vorwiegend angewandten CERAD- und NIH-RI-Kriterien modifizierten „ABC“-Score für die histopathologische Erfassung von β-Amyloid (A), Stadieneinteilung der Neurofibrillendegeneration (B) und Erfassung neuritischer Plaques (C) für die Beurteilung, mit welcher Wahrscheinlichkeit eine Demenz durch Alzheimer-typische Veränderungen bedingt sei;
- den Vorschlag für eine detaillierte Erfassung häufiger komorbider Veränderungen, wie DLB, vaskuläre Hirnschäden, Hippokampussklerose und TDP-43 immunreaktiver Einschlüsse.
Ferner sind in der Diagnostik verschiedene klinisch relevante Untergruppen der AD und die im fortgeschrittenen Alter zunehmenden Komorbiditäten und Begleitpathologien zu berücksichtigen.
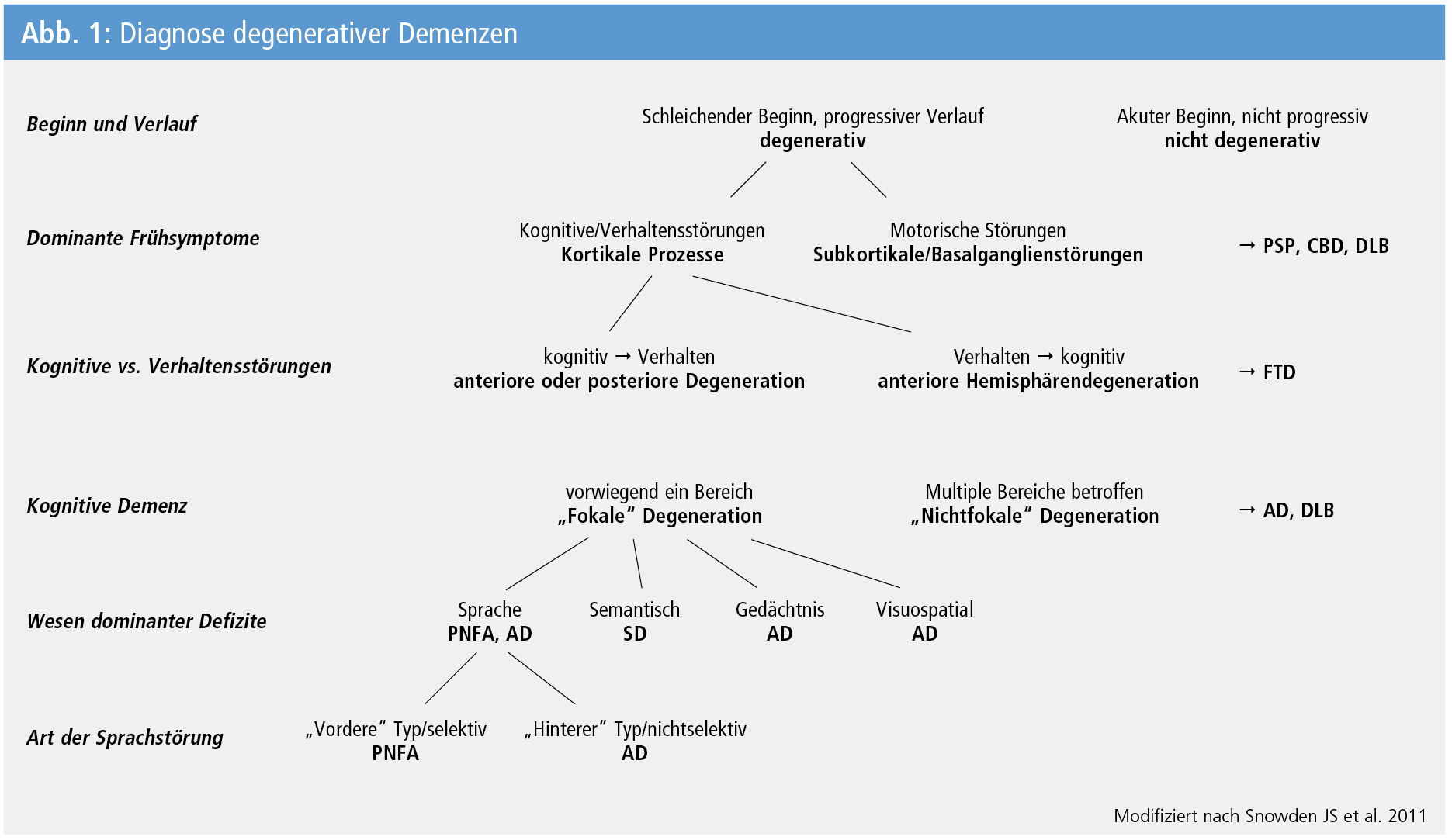
Einen Algorithmus für die Diagnose degenerativer Demenzen zeigt Abbildung 1.
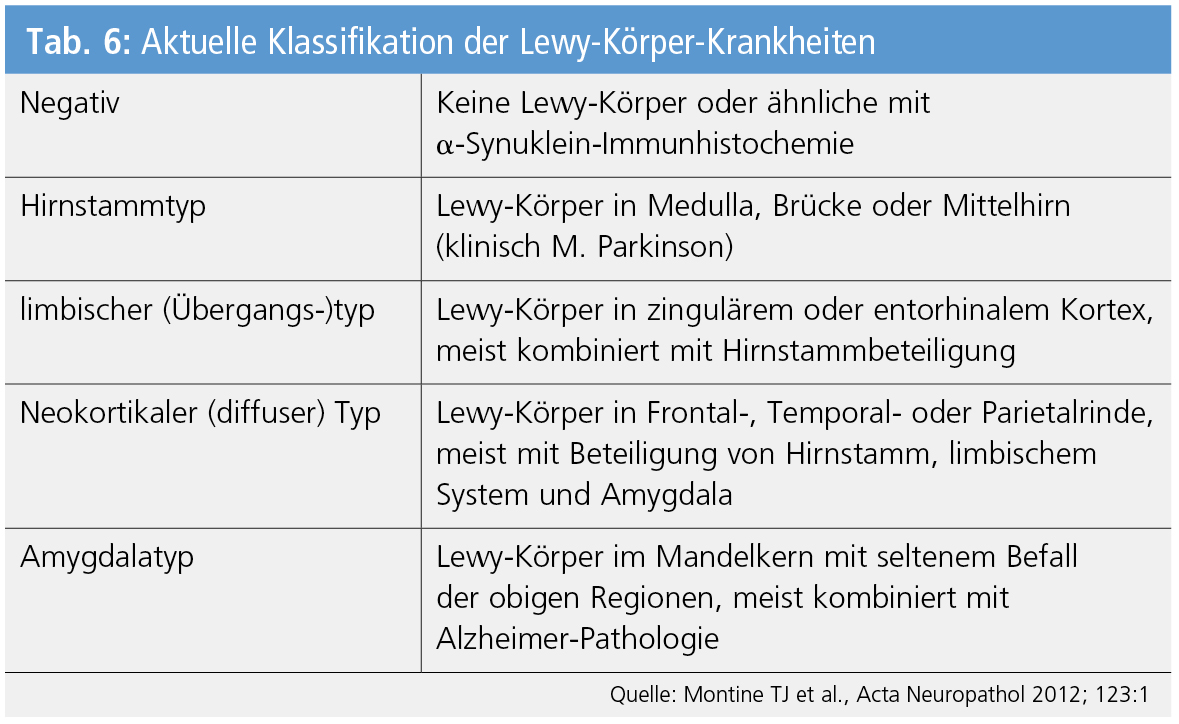
Bei Synukleinopathien, insbesondere Lewy-Körper-Prozessen (DLB, M. Parkinson, PDD) besteht ein Befall weiter Anteile des zentralen, autonomen und peripheren Nervensystems sowie multipler Organe durch phosphoryliertes α-Synuklein mit frühester Beteiligung des Gastrointestinaltraktes und des Bulbus olfactorius. Die durch die Einteilung der Lewy-Pathologie in 6 Stadien mit Progression von der Medulla oblongata zur Großhirnrinde für den Krankheitsverlauf vermutete Bedeutung wird derzeit eifrig diskutiert, was zu einer modifizierten Klassifikation der Lewy-Körper-Krankheiten führte (Tab. 6).
Für die frontotemporalen Demenzen oder Frontotemporallappen-Degeneration (FTLD) liegen aktuelle Klassifikationskriterien vor, welche die verschiedenen molekularen Marker berücksichtigen (Abb. 2), wobei allerdings häufige Überlappungen untereinander und mit der DLB bestehen. Die FTLD bei älteren Personen bietet gleichfalls verschiedene Phänotypen und morphologische Untergruppen ähnlich wie bei jüngeren PatientInnen, wobei MAPT(Tau-Gen)-Mutationen und FUS-Pathologie wesentlich seltener sind oder fehlen.

Für die vaskulären Demenzen bestehen verschiedene Kategorisierungsvorschläge, etwa die revidierten NIA-AA-Richtlinien zur Erfassung aller makroskopischen und mikroskopischen vaskulären Hirnläsionen und ihren Zusammenhang mit kognitiven Einbußen oder ein jüngster, komplizierter Vorschlag zur semiquantitativen Erfassung zerebrovaskulärer Läsionen in 6 Stadien. Wichtig ist eine Harmonisierung der Kriterien und morphologischen Untersuchungstechniken zur Erfassung vaskulär-ischämischer Hirnschäden und ihrer Beziehung zu kognitiven Einbußen.
Spezielle diagnostische Probleme
Die zunehmende Anwendung biochemischer, genetischer und experimenteller Methoden hat eine Verfeinerung der Diagnose und Analyse der zu Demenz führenden relevanten Prozesse bewirkt. Da die Mehrzahl degenerativer Demenzen mit der Ablagerung pathogener Proteine als krankheitstypischen Markern einhergeht, ist deren Nachweis unter Anwendung moderner molekular-pathologischer Kriterien ein wichtiger diagnostischer Zugang. Wegen der häufigen Überlappungen zwischen kognitiv intakten SeniorInnen, solchen mit MCI oder Frühstadien von Demenzen und deren Vollbild ist ihre diagnostische Abgrenzung oft schwierig. Zwar ist nur ein geringer Teil kognitiv intakter älterer Menschen frei von pathologischen Hirnläsionen, doch zeigen bis zu 50 % kognitiv unauffälliger SeniorInnen Alzheimer-typische oder andere Läsionen, die synergistische oder additive Effekte auf die Hirnleistung haben.
Die moderne Neuropathologie erzielt bei Anwendung aktueller Methoden eine Diagnose oder Klassifikation in bis zu 99 % der neurodegenerativen Demenzen, ohne jedoch mit Ausnahme von Fällen mit gesichertem genetischen oder metabolischen Hintergrund ihre Ursachen (Ätiologie) klären zu können. Aktueller Kenntnisstand und künftige interdisziplinäre Forschung sollen einen Weg aus dem „Chaos“ in der Diagnostik dementiver Prozesse und ihrer klinischen Implikationen als Voraussetzungen für eine erfolgreiche Prävention und Therapie dieser deletären und bisher nur symptomatisch behandelbaren Erkrankungen zeigen.
Literatur beim Verfasser

AutorIn: Univ.-Prof. Dr. Kurt Jellinger
Institut für Klinische Neurobiologie, Wien

Ursprünglich erschienen:
neuro 04|2012
neuro 04|2012

