
























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Am Beispiel des Nierenzellkarzinoms: Epigenetische Modulation mit Histon-Deacetylase-Inhibitoren
25. Februar 2011
Derzeit sind folgende Therapiemodalitäten für das metastasierte Nierenzellkarzinom zugelassen: Sunitinib, Bevacizumab plus IFN, Temsirolimus und Pazopanib (Multikinaseinhibitor gegen VEGFR-1-3, PDGFR-α, -β und c-Kit) als First-Line-Therapie sowie Sorafenib und Everolimus als Second-Line- Therapie.
Therapien mit Multikinasehemmern sind beim metastasierten Nierenzellkarzinom auf evidenzbasierter Basis wirksam und führen auch als Individualtherapie bei nichtselektionierten PatientInnen zu anhaltenden Tumorremissionen. Dennoch ist auch bei diesen neuen zielgerichteten Therapien die Resistenzentwicklung ein Problem, das den klinischen Alltag bestimmt.
Daher ist die Suche nach neuen Pathways und Signalwegen, die für Proliferation und Ausbreitung sowie für die Metastasierung des Nierenzellkarzinoms elementar sind, von essenzieller Bedeutung. Eine Unterbrechung bzw. Blockade dieser Pathways sollte zu einem Verlust der proliferationsfördernden Eigenschaften von Krebszellen und damit zu einer Hemmung des Wachstums führen.
Interessante Kandidaten dafür werden auf Basis ihres Wirkmechanismus als „epigenetische Modulatoren“ in einer Substanzgruppe zusammengefasst. Wichtige Vertreter sind die DNS-Methyltransferase- Inhibitoren (DNMTI) und die Histon-Deacetylase-Inhibitoren (HDACInhibitoren). In diesem kurzen Überblick wird ausschließlich auf die Klasse der HDAC-Inhibitoren eingegangen und der Bezug dieser Therapeutika zum Nierenzellkarzinom dargestellt.
Charakteristika der epigenetischen Vererbung
Die Epigenetik beschäftigt sich mit der epigenetischen Vererbung, d.h. der Weitergabe von Eigenschaften auf die Nachkommen, die nicht auf Veränderungen in der DNA-Sequenz zurückgehen, sondern auf hereditären Änderungen der Genregulation und Genexpression beruhen. Epigenetische Prozesse beeinflussen die Genregulation durch Veränderungen von Struktur und Konformation der DNS innerhalb des Chromatins. DNS-Methylierung und Histon-Deacetylierung sind zwei Beispiele für epigenetische Prozesse bzw. epigenetische Modifikationen.
Modifikation von Histonen
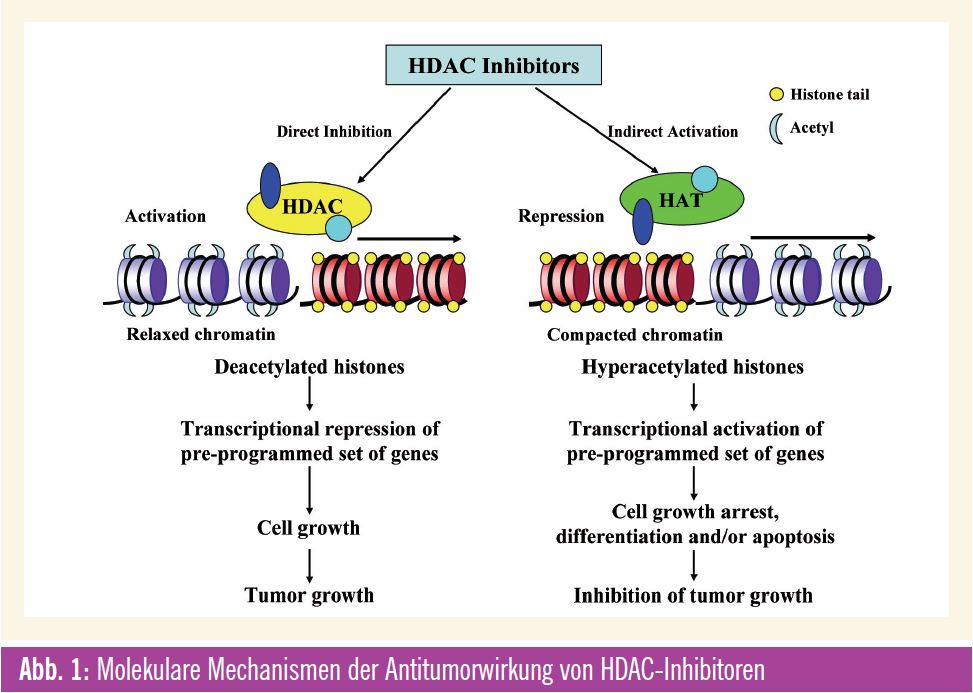
Histone sind Proteine, die im Zellkern von Eukaryonten vorkommen. Sie sind als Bestandteil des Chromatins für die Verpackung der DNA, aber auch für die Expression mancher der auf ihr codierten Gene von essenzieller Bedeutung. Eine Modifikationen von Histonen führt zu einer stärkeren oder schwächeren Verpakkung des Chromatins und damit zur Aktivierung bzw. Stilllegung (Gen-Silencing) umliegender DNA-Abschnitte. Die Modifikation der Histone ist mit der Transkriptionsaktivierung gekoppelt und kann auch nach Zellteilung bestehen bleiben. Histon-Acetylierung wird durch die sog. Histon-Acetyltransferasen (HAT) mediiert, während die Entfernung der Acetylgruppen durch Histon-Deacetylasen (HDAC) erfolgt. Die Familie der HDACProteine besteht aus 18 Isoformen, die in 4 Klassen eingeteilt werden. Durch Veränderungen der DNS oder von strukturellen Komponenten des Chromatins wird die Expression von Suppressorgenen und die Aktivität von Transkriptionsfaktoren, die bei der Initiation und Progression von Krebs beteiligt sind, reguliert und moduliert.
HDAC-Expression in physiologischem vs. malignem Gewebe
Sowohl bei hämatologischen Malignomen (Lymphome; kutanes T-Zell-Lymphom) als auch bei soliden Tumoren (Magen-, Kolorektal-, Ovarial-; Endometrium-, Leber-, Pankreas-, nichtkleinzelliges Lungen- und auch Mammakarzinom) ist eine hohe Expression von HDAC, vorwiegend Isoenzyme der Klasse I (HDAC 1,2,3), nachgewiesen worden. Dabei ist eine dementsprechende Hypo- Acetylierung von Histonen ein häufiger Befund. Beim Vergleich von normalem mit malignem Gewebe wird ein höherer Aectylierungslevel im normalen lymphatischen Gewebe als etwa im Lymphom festgestellt. Gleiches gilt für Dickdarmgewebe im Vergleich zum Kolonkarzinom: So ist auch die HDAC1-Enzymexpression im Adenokarzinom höher als im normalen Kolonepithel. Der Verlust einer genetischen „Gatekeeper-Funktion“ in präkanzerösen Läsionen kann mit einer verstärkten HDAC-Aktivität einhergehen, wie das z.B. bei der Kolon-Kanzerogenese festgestellt wurde.
HDAC-Expression und -Inhibition beim Nierenzellkarzinom: Kürzlich konnte in einer Studie gezeigt werden, daß HDAC auch beim Nierenzellkarzinom vermehrt vorhanden sind. So war gezeigt worden, dass nahezu 60 % der Nierenzellkarzinome die Isoform 1 und 2 sowie etwa 13 % der Nierenzellkarzinome die Isoform 3 exprimieren. Bei der Entstehung des Nierenzellkarzinoms ist der Funktionsverlust des Von-Hippel-Lindau-Suppressor- Gens (VHL) dokumentiert. Dadurch kommt es zu einer vermehrten Expression von HIF1-regulierten Genen und damit zur verstärkten Neo-Angiogenese. Eine epigenetische Modifikation von Genen durch erhöhte HDAC-Level und eine vermehrte Deacetylierung von Histonen kann ein Mechanismus sein, der zur Stilllegung von Suppressor-Genen und anderen Genen führt, die für Zellzyklusprogression, Zellproliferation, Differenzierung und Apoptose verantwortlich sind.
Angriffspunkte für HDAC-Inhibitoren
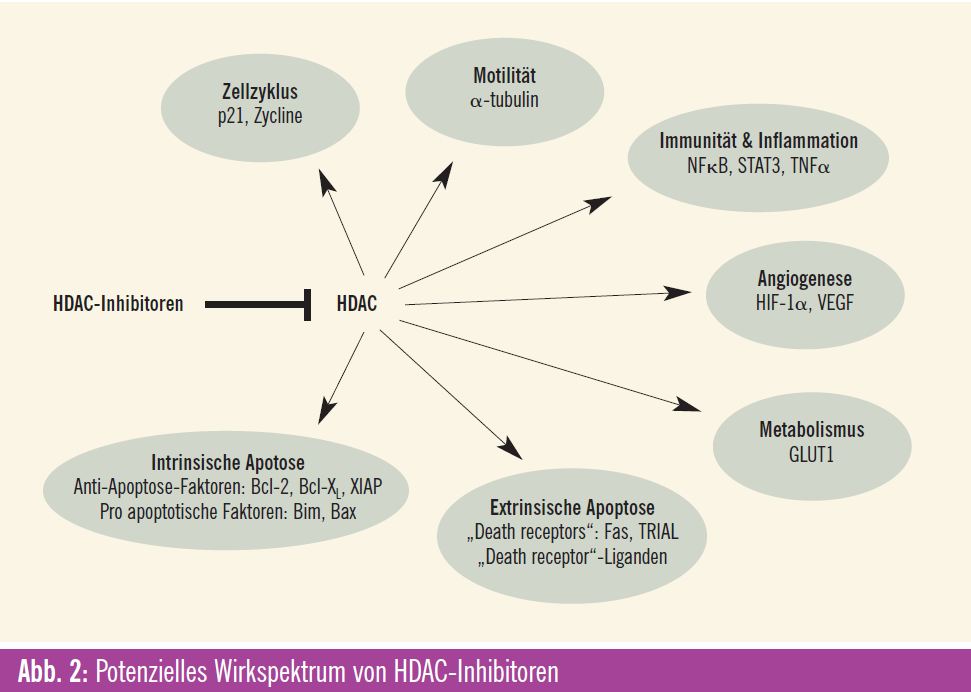
Die epigenetische Modifikation von Histonen ist ein Angriffspunkt für die Entwicklung von neuen Krebstherapien, wobei gerade den HDAC-Inhibitoren als epigenetischen Modulatoren große Bedeutung zugemessen wird. Der Wirkeffekt dieser immer größer werdenden Gruppe von HDAC-Inhibitoren liegt primär in der Induktion einer Histon-Acetylierung, wodurch eine offene Konfiguration des Chromatins ermöglicht wird und eine Reaktivierung von supprimierten Genen stattfindet. Das Resultat mani – festiert sich für Krebszellen als Zell – zyklusarrest, Zelldifferenzierung und Apoptose. Für die verschiedenen HDACInhibitoren ist das Muster für die Veränderungen der Genexpression gleich, aber die Auswirkungen sind unterschiedlich. Zurzeit sind mehr als 18 HDAC-Inhibitoren in präklinischer Entwicklung bzw. werden sie bereits in klinischen Phase-I-, -II- und -III-Studien bei verschiedensten malignen Erkrankungen untersucht. Der allgemeine Wirkmechanismus vieler dieser Medikamente ist eine Bindung an das Zn2+-Ion, das für die katalytische Funktion der HDAC-Enzyme notwendig ist, wodurch diese Reaktion gehemmt wird. Wiewohl diese Stoffklasse auf Basis der Eigenschaft HDAC zu binden, entwickelt wurde, haben einzelne Verbindungen weitere und durchaus unterschiedliche Wirkungen. Besonders die variablen Effekte auf die (Hyper-) Acetylierung von „Non-Histon“-Proteinen/ Substraten (wie HSP90, Raf, Akt, ErB2 und BCR-ABL) können auch wesentlich zur antitumoralen Wirkung beitragen. Diese transkriptionalen und nichttranskriptionalen Effekte der HDACInhibitoren kennzeichnen diese Wirkstoffklasse. Es handelt sich somit um Medikamente, die multiple Pathways und Signalwege beeinflussen können, wodurch sie sich als sehr attraktive neue zielgerichtete Therapien für den klinischen Einsatz bei malignen Erkrankungen qualifizieren.
Vorinostat (Suberoylanilide Hydroxamsäure, SAHA) wurde als erstes Medikament dieser Klasse bereits 2006 von der FDA für die Behandlung des kutanen T-Zell-Lymphoms (CTCL) für Patienten mit Progression oder Rückfall der Erkrankung nach 2 systemischen Therapien zugelassen. Die Therapie wird oral verabreicht (400 mg/Tag) und ist relativ gut verträglich. In der Zulassungsstudie wurde eine objektive Ansprechrate von 30 % erzielt. Zahlreiche Phase-II- und -III-Studien sind derzeit in Durchführung, wobei eine Wirksamkeit auch bei anderen Lymphomen dokumentiert wurde.
Beim Nierenzellkarzinom wurde die Monotherapie mit HDAC-Inhibitoren mit Ausnahme von Phase-I-Studien klinisch noch nicht intensiv getestet, wiewohl es Argumente gibt, die für einen solchen Einsatz sprechen: So ist der Nachweis der Expression von HDAC bei 60 % der Nierenzellkarzinome nachgewiesen, auch der Verlust der Funktion des VHLSuppressor- Gens und besonders auch die Wirkung der Substanzklasse auf Non-Histon-Proteine.
Die Kombination von HDAC-Inhibitoren mit anderen Therapien wie Chemound/ oder Strahlentherapie, aber auch mit neuen zielgerichteten Therapien ist präklinisch mit guten Ergebnissen getestet worden. Klinische Phase-I-Studien bei fortgeschrittenen soliden Tumoren haben die gute Verträglichkeit von Vorinostat mit verschiedenen Chemotherapien wie Pemetrexed, Cisplatin, Doxorubicin, 5FU/LV und Oxaliplatin, Carboplatin plus Paclitaxel, Capecitabin und auch Temozolomid dokumentiert und mögliche Dosierungen für Phase-II-Studien bestimmt.
Eine rezente präklischen Studie an In-vitro- und In-vivo-Modellen des Nierenzellkarzinoms hat dokumentiert, dass die dabei nachgewiesene antineoplastische Wirkung der Kombination von Vorinostat mit dem m-TOR-Inhibitor Temsirolimus auf vielen Ebenen stattfindet. Eine starke Reduktion der Survivin-Spiegel ist von einer Induktion der Apoptose und einer Hemmung der Tumorzellproliferation gefolgt. Weiters wurde mit der Kombination ein stärkerer Hemmeffekt auf die Angiogenese im Vergleich zu den Monosubstanzen festgestellt: Diese Ergebnissen mit diesen In-vitro- und In-vivo-Modellen des Nierenzellkarzinoms lassen die Schlussfolgerung zu, dass Vorinostat die antineoplastische Wirkung von Temsirolimus verstärkt.
ZUSAMMENFASSEND ist festzustellen, dass die epigenetische Modulation durch HDAC-Inhibitoren – wie es hier beispielhaft dargestellt wurde – durchaus ein gültiges Therapiekonzept für das Nierenzellkarzinom sein kann, das aber durch Ergebnisse von klinischen Studien bestätigt werden muss. Interessante Mög lichkeiten in diesem Kontext bieten sich besonders auch in der Kombinationstherapie bzw. nach Versagen von zielgerichteten Therapien mit Multikinaseinhibitoren zur Überwindung der Therapieresistenz.

Ursprünglich erschienen:
SO 01|2011
SO 01|2011
Herausgeber: Univ.-Prof. Dr. Christoph Zielinksi, Univ.-Prof. Dr. Markus Raderer
Publikationsdatum: 2011-02-25
Zur Ausgabe »
Publikationsdatum: 2011-02-25
Zur Ausgabe »

