
























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Diagnose, Prognose und JAK2-Hemmer als neue Therapieoption – Myeloproliferative Erkrankungen
26. August 2011
Diagnose, Prognose
Nachdem die Knochenmarksbiopsie im Jahr 2001 von der WHO als wesentliches Kriterium zur Diagnose myeloproliferativer Erkrankungen aufgenommen wurde, setzte die JAK2-Mutationstestung im Jahr 2008 einen neuen diagnostischen Meilenstein (Tefferi A et al., Blood 2007; 110:1092 ff.). Die Häufigkeit einer JAK2-V617F-Mutation betrug in einer Studie von Vannucchi et al. bei Polycythaemia vera etwa 98 %, bei essenzieller Thrombozythämie rund 60 %, bei primärer Myelofibrose 62 % und bei sekundärer Myelofibrose 82 % (Vannucchi AM et al., Blood 2007; 110:840 ff.). Bei JAK2-V617F-negativen PV-Patienten fanden sich häufig andere Mutationen, etwa JAK2-Exon-12-Mutationen (in einer Größenordnung von 40 % in der gleichen Studie von Vannucchi et al.). Seit 2008 beinhalten die wesentlichen Diagnosekriterien der Polycythaemia vera einen Hämoglobinwert > 18,5 g/dl bei Männern, > 16,5 g/dl bei Frauen und das Vorliegen einer JAK2-Mutation. Beide Kriterien gemeinsam und ein zusätzliches Kriterium (z. B. niedriges Serumery – thropoietin) sichern die Diagnose. Im Weiteren ging der Vortrag von Guido Finazzi auf die Notwendigkeit der Knochenmarkbiopsie zur Unterscheidung der frühen/präfibrotischen Myelofibrose (hyperzelluläres Bild mit Megakaryozyten- Clustern, chromatinreichen Nuklei und proliferierenden Granulozyten) von der essenziellen Thrombozythämie (normozelluläres Bild mit verstreuten Megakaryozyten) ein. Hintergrund der Diskussion ist eine rezent im JCO publizierte Studie von Barbui et al., die der Frage nachging, ob die Unterscheidung der ET von der frühen PMF durch die strikte Anwendung der WHO-Kriterien klinisch und prognostisch relevant ist (Barbui T. et al., J Clin Oncol 2011; 29:3179 ff.). Die Studie inkludierte 1104 Patienten, bei denen zwischen 1975 und 2008 die Diagnose ET gestellt wurde und von denen diagnostische (prätherapeutische) Knochenmarkbiopsien verfügbar waren. Nach neuer Begutachtung der verfügbaren Samples im Einklang mit den WHOKriterien wurde bei 891 Patienten (81 %) die Diagnose ET bestätigt und bei 180 Patienten (16 %) auf frühe PMF revidiert (3 % nicht evaluierbar). Mit einem Follow-up von 6–7 Jahren zeigte sich aus klinischer Sicht zwischen beiden Patientengruppen ein ähnlicher Verlauf, was die Inzidenz von Thrombosen betrifft (die zu den häufigsten Todesur – sachen beider Erkrankungen zählen). Signifikant unterschiedlich war (erwartungsgemäß) die Evolution der frühen/ präfibrotischen PMF in das Vollbild der Myelofibrose (gegenüber ET-Patienten doppelt so häufig, p = 0,04) und die Transformation in eine akute Leukämie (AML): Das AML-Risiko war bei Patienten mit früher PMF 5,2-mal so hoch als bei ET-Patienten (p = 0,0012). Schließlich hatten Patienten mit früher PMF ein doppelt so hohes Mortalitätsrisiko (22 % vs. 10 %, p = 0,0002). Als wesentliche Risikofaktoren für die Entwicklung einer Thrombose wurden bereits stattgefundene Thrombosen und ein Patientenalter _ 65 Jahre für beide Erkrankungen bestätigt (Marchioli R et al., J Clin Oncol 2005; 23:2224 ff.). Abschließend wurde auf die Notwendigkeit der prospektiven Validierung neuer oder noch nicht im IPSS-Scoring-System enthaltener Thrombose- Marker eingegangen. So scheint die Leukozytose ein bedeutender Risikofaktor für venöse und arterielle Thrombosen, Myokardinfarkte und Schlaganfälle bei ET-Patienten zu sein (Carobbio A et al., J Clin Oncol 2008; 26:2732 ff). Darüber hinaus waren JAK2-Mutationen bei essenzieller Thrombozythämie und bei primärer Myelofibrose mit einer doppelt bis dreifach so hohen Inzidenz von thromboseassoziierten Komplikationen vergesellschaftet (Finazzi G et al., Haematologica 2007; 92:135 ff.).
JAK-1/2-Hemmer als neue Therapieoption
Von Serge Verstovsek wurde festgehalten, dass trotz einer Reihe individueller Managementstrategien (Transplantation, Anämiebehandlung, Therapie der Splenomegalie, Strahlentherapie, Supportivmaßnahmen zur Erhaltung der Lebensqualität) bis zur Etablierung von JAK2- Inhibitoren keine spezifische Therapie bei Myelofibrose verfügbar war, die sich auf evidenzbasierter Basis qualifiziert hätte. Es sind allerdings etliche neue oder aus anderen Indikationen bekannte Therapieansätze in klinischer Erprobung (immunmodulatorische und hypomethylierende Substanzen, Proteasomeninhibitoren, Tyrosinkinasehemmer, Angiogenesehemmer oder mTOR- und Histonde – acetylase-Inhibitoren), wenngleich etliche der bisherigen Studien negativ verlaufen sind und nur wenige Substanzen weiterverfolgt werden – laut Serge Verstovsek vor allem mTOR- und Histondeacetylase- Inhibitoren als Monotherapie oder in Kombination mit JAK2- Hemmern.
In den JAK-STAT-Signaling- Pathway sind vier Mitglieder der JAK-Familie involviert (JAK1–3 und Tyk2), wobei die JAK2-Tyrosinkinase erythropoietinund thrombopoietinassoziierte Wachstumsfaktorsignale für die normale Hämatopoese vermittelt. Die JAK2-V617F-Mutation wurde vor 6 Jahren in multipotenten Progenitorzellen entdeckt und führte – als proof of concept für neue Therapien – über die konstitutive Aktivierung der JAK2-Kinase zu Polycythaemia vera → Myelofibrose im Tiermodell. Heute weiß man, dass die Mutation zwar bei einem Großteil der Patienten mit ET, MF und PV nachweisbar ist und zur Erkrankung beiträgt, aber nicht kausal damit verknüpft ist. Das heißt, unabhängig vom Mutationsstatus gilt die Dysregulation im JAK-Pathway als Schlüssel für myeloproliferative Erkrankungen (Quintas Cardama A, Nat Rev Drug Discov. 2011; 10:127 ff). Wesentlich für die Therapie mit JAK2-Inhibitoren und Ruxolitinib als dem am weitesten entwickelten Medikament dieser neuen Substanzklasse ist das Wissen, dass Januskinaseinhibitoren die ATPBindungsstelle hemmen und daher nicht selektiv bei mutierten JAK-Varianten zur Wirkung kommen, sondern gleichzeitig auch bei Wildtyp-Patienten (allerdings gegenüber ausgewählten Mutationen resistent sein können) u. U. präferenziell auf Zellen wirken, die eine Mutation tragen und damit die konstitutiv aktivierte Kinase aufweisen, dass Myelosuppression eine erwartete Nebenwirkung dieser Substanzklasse ist und dass es unwahrscheinlich ist, dass die Erkrankung mit JAK2-Hemmern eradiziert wird (da Patienten bei höherer Dosis einer Aplasie erliegen würden). Die Wirksamkeit von JAK2-Inhibitoren bei Myelofibrose wurde anhand jener Kriterien untersucht, die klinisch am meisten ins Gewicht fallen: Zytopenie (Anämie), Splenomegalie und Lebensqualität (Performance Status) – deren Verbesserung gegenüber der myelosuppressiven Wirkung abzuwägen wäre. Sieben JAK2-Inhibitoren sind in früher klinischer Entwicklung (Phase I, II), mit Ruxolitinib sind zwei Phase-III-Studien bei MF-Patienten bereits abgeschlossen und eine Phase-III-Studie bei PV-Patienten vor kurzem initiiert worden.
Als Update zu Ruxolitinib wurden neue Daten aus der Phase I, II Studie im New England Journal of Medicine publiziert wurde (Verstovsek S et al., N Engl J Med 2010; 363[12]:1117 ff.) präsentiert, die bei 157 MF-Patienten eine Dosis von 25 mg zweimal täglich (BID) als maximal verträgliche Dosis (MTD) evaluierte (Thrombozytopenie war die dosislimitie – rende Toxizität) und die Therapie je nach Sicherheit und Verträglichkeit individuell optimierte (Phase-I/IIStudie). Ruxolitinib wurde be vor zugt mit 15 mg BID (oder 20 mg BID bei einer Plättchenzahl > 200.000/μl) be gonnen, womit hämatologische Nebenwirkungen (Thrombozythämie, Anä mie) gering gehalten wurden; eine erste Steigerung auf 20 mg BID erfolgte nach 1 Monat (bei inadäquatem Ansprechen und guter Verträglichkeit, vor allem gemessen an Splenomegalie und Lebensqualität), und eine weitere Steigerung auf 25 mg BID erfolgte nach 2 Monaten, wenn das Ansprechen bei guter Verträglichkeit immer noch inadäquat erschien. Bei einer Plättchenzahl < 100.000/μl sollte die Dosis reduziert werden. Laut Verstovsek lässt sich die individuell optimale Dosis innerhalb von 3 Monaten finden und kann dann beibehalten werden. Insgesamt befinden sich 115/157 Patienten (73 %) in verschiedenen Dosisstufen über einen medianen Zeitraum von 19,4 Monaten immer noch in der Studie. Nach 19 Monaten Followup betrug die Inzidenz einer Grad-3- Thrombozytopenie in der Dosis von 15 bzw. 25 mg BID jeweils 3 % (1 Patient) bzw. 26 % (12 Patienten) und die In – zidenz einer Grad-4-Thrombozythämie 0 % bzw. 11 % (5 Patienten). Nichthämatologische Nebenwirkungen Grad 3 lagen insgesamt zwischen 0 und 2 %, Grad-4-Nebenwirkungen wurden nicht verzeichnet. Als primärer Wirksamkeitsparameter konnte die palpable Milzgröße bei den meisten Patienten um 25 % oder um 50–100 % reduziert werden, und zwar unabhängig vom Vorliegen einer JAK-Mutation (Abb. 1).
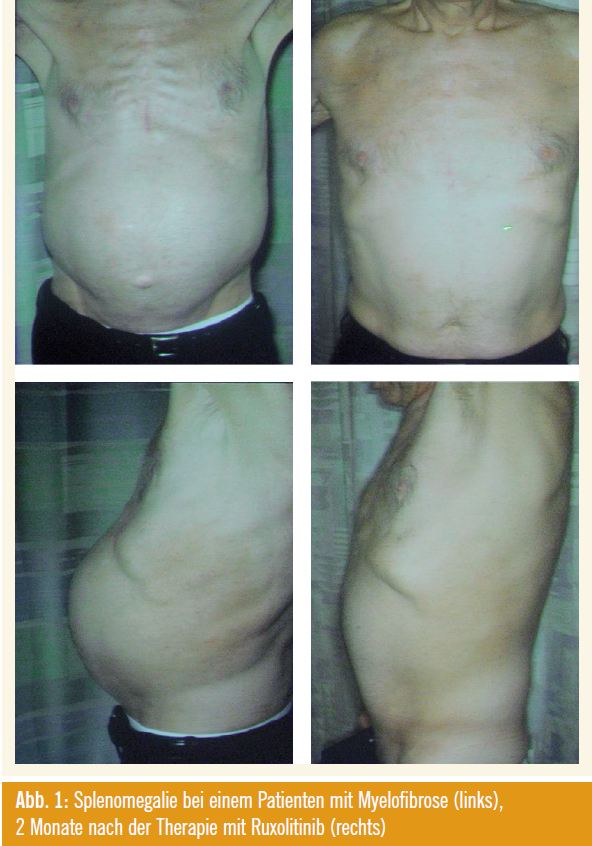
Dabei korrelierte ein Rückgang der palpablen Milzgröße um 50 % mit einem MRI-gemessenen Rückgang um 35 %. Insgesamt wurde festgehalten, dass die Patienten sehr rasch ansprachen und der Respons über den Beobachtungszeitraum anhielt. Darüber hinaus konnten anhand eines gemeinsam mit der FDA neu entwickelten Myelofibrosis Symptom Assessement Formulars (MSFAF), das die 10 wichtigs ten Symptome der Patienten beinhaltet, insbesondere abdominelle Schmerzen, Juckreiz, Nachtschweiß, Knochen- oder Muskelschmerzen um zumindest 50 % reduziert werden. Interessanterweise korrelierte der Rückgang der Symptome weniger mit dem Rückgang der Milzgröße als mit einer dauerhaften Suppression inflammatorischer Plasmazytokine (C-reaktives Protein, IL-1ra, MP16, TNF-alpha, IL-6). Myelofibrose-Patienten, die im 6-Minuten- Gehtest schlechter abschneiden als altersgematchte gesunde Personen, profitierten über den Therapiezeitraum von einer deutlichen Verbesserung ihrer Belastbarkeit. Als Wirkung auf das Blutbild und das Knochenmark wurde festgehalten, dass Leukozyten und Thrombozyten auf normale Werte zurückgingen und dass einige Patienten von einer lang anhaltenden Transfusionsunabhängigkeit profitierten, dass JAK2-V617-Allele zurückgehen können, während Blasten und Knochenmarkfibrose unter der Therapie auf lange Zeit stabil bleiben und der Therapieerfolg hier als Krankheitsstabilisierung zu werten ist.
COMFORT I (USA, Kanada, Australien), COMFORT II (Europa): In den beiden Phase-III-Zulassungsstudien, deren Rekrutierung abgeschlossen ist, wurde Ruxolitinib mit einer Startdosis von 15 oder 20 mg BID gegen Placebo oder (in Europa) gegen die beste verfügbare Therapie untersucht; primärer Endpunkt war ein Rückgang der Milzgröße um 35 % innerhalb von 24 oder (in Europa) 48 Wochen gemessen mit MRI.
In der europäischen Zulassungsstudie COMFORT II konnte Ruxolitinib nach 48 Wochen bei 28,5 % der Patienten mit intermediärem- 2 oder hohem Risiko (nach IPSS) eine Reduktion der Milzgröße um zumindest 35 % erreichen (vs. 0 % mit bester verfügbarer Therapie; p < 0,0001) (Abb. 2). Nach 24 Wochen (sekundärer Studienendpunkt) war dies bei 31,9 % der Patienten der Fall (vs. 0 % im Vergleichsarm). Zwei Drittel der Patienten im Vergleichsarm erhielten zumindest ein oder mehrere Medikamente (antineoplastische Substanzen, Hydroxyurea, Glukokortikoide, Epoetine, Immunmodulatoren u. a.), ein Drittel erhielt keine Supportivtherapie. Die Möglichkeit eines Cross-overs auf Ruxolitinib war im Studiendesign vorgesehen und wurde bereits nach kurzer Zeit von einem Viertel der Patienten in Anspruch genommen. Patienten im Ruxolitinib-Arm sprachen rasch auf die Therapie an (innerhalb von median 12 Wochen, d. h. bereits zum Zeitpunkt der ersten Evaluierung), wobei der Therapieerfolg über den Beobachtungszeitraum bei den meisten Patienten aufrecht blieb. Deutliche Unterschiede ergaben sich auch in verschiedenen Instrumenten zur Evaluierung der Lebensqualität (speziell auch in Hinsicht auf Appetit, Dyspnoe, Schmerzen, Schwindelgefühl oder Fatigue). Die hauptsächlichen Target-assoziierten Nebenwirkungen sind Anämie und Thrombozytopenie, die bereits mit Studienbeginn bei den meisten Patienten in leichter Form vorhanden waren. Nichthämatologische Nebenwirkungen traten im Therapieverlauf mehrheitlich als Grad-1/2-Nebenwirkung auf.
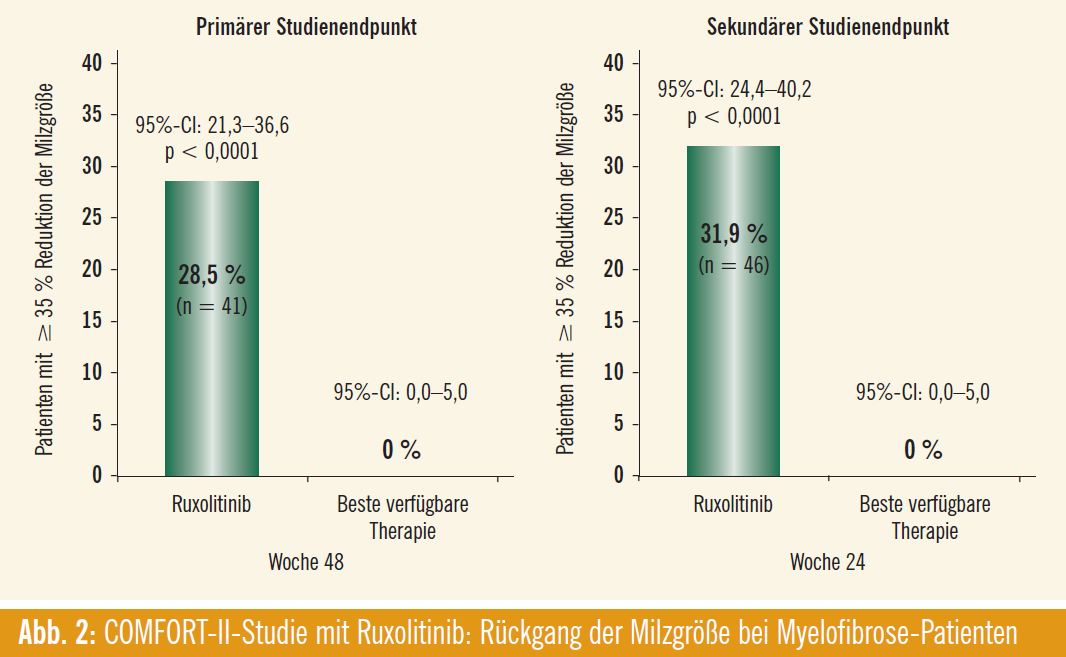
Im Fazit wurde festgehalten, dass Ruxolitinib in der COMFORT-II-Studie ähnlich wie in der COMFORT-I-Studie einen deutlichen, rasch einsetzenden und dauerhaften klinischen Benefit bei Myelofibrose- Patienten unabhängig vom Vorliegen einer JAK2-V617F-Mutation erzielte und sich als wertvolle neue Therapieoption in dieser Indikation anbietet.
Man kann ausblickend darauf hinweisen, dass Ruxolitinib auch bei Polycythaemia vera in einer Phase-II-Pilotstudie erfolgreich getestet wurde und – gemessen an den Ansprechkriterien des European Leukemia Net (Hämatokrit, Plättchencount, weiße Blutzellen, Milzgröße, krankheitsassoziierte Symptome) – partielle und komplette Remissionen bei jeweils 50 % der Patienten erzielte und auf dieser Basis in einer Phase-III-Studie (RESPONSE) gegen Hydroxyurea weiter evaluiert wird.
*Quelle: EHA-Symposium und Presidential-Symposium: „Evolving Paradigms in the Diagnosis and Prognosis of Myeloproliferative Neoplasms“, Guido Finazzi (Ospedale Riuniti, Bergamo); „Myeloproliferative Neoplasms: Current and Emerging Therapies“, Serge Verstovsek (MD Anderson Cancer Center, Houston)

Ursprünglich erschienen:
SO 03|2011
SO 03|2011
Herausgeber: Univ.-Prof. Dr. Christoph Zielinksi, Univ.-Prof. Dr. Markus Raderer
Publikationsdatum: 2011-08-26
Zur Ausgabe »
Publikationsdatum: 2011-08-26
Zur Ausgabe »
