





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Paradigmenwechsel von der Klonalität zur Tumorstammzelle?
23. Dezember 2011
Basierend auf dem Modell der klonalen Evolution und der Annahme, dass die meisten Tumorzellen das Potenzial zur Proliferation und Selbsterneuerung haben, war es das Ziel der traditionellen Krebsbehandlung, möglichst viele – idealerweise alle – Tumorzellen zu vernichten. Im Gegensatz müsste es entsprechend der Tumorstammzellhypothese genügen, dass nur wenige Tumorzellen vernichtet werden, nämlich jene Stammzellen (CSC = cancer stem cells), welche für das Wachstum, die Selbsterneuerung und die Persistenz einer Krebserkrankung verantwortlich sind. CSC wurden mittlerweile bei den meisten soliden Tumoren und Leukämien aufgrund ihrer Repopulationsfähigkeit in immunsupprimierten Mäusen nachgewiesen. Die therapeutische Konsequenz hat zur Entwicklung von fokussierten Therapiekonzepten geführt, welche auf die gezielte Vernichtung der CSC abzielen, die eine traditionelle Behandlung möglicherweise überstehen, was für den relativ schlechten Therapieerfolg bei vielen aggressiven Krebserkrankungen verantwortlich ist.
Angesichts einer steigenden Zahl von experimentellen Befunden hat das CSCKonzept in den vergangenen 15 Jahren zunehmend an Bedeutung gewonnen. Obwohl man ursprünglich angenommen hat, dass nur eine relativ kleine Population von Stammzellen einerseits die Fähigkeit zur Selbsterneuerung hat und andererseits verschiedenste Zellpopulationen innerhalb eines Malignoms hervorbringen kann, haben neuere Publikationen gezeigt, dass diese Zellen – zumindest in einigen Malignomen – gar nicht so selten sein müssen. Kritische Auseinandersetzungen mit der Interpretation diverser CSC-Marker bilden den Nährboden für eine ständige Optimierung des CSC-Modells.
Grundsätzlich gehen beide Modelle von Differenzierungssystemen aus, bei denen – ausgehend von Stammzellen – eine Reihe von Vorstufenzellen entstehen und letztlich ausgereifte Zellen hervorgehen.
Die Tumorstammzellhypothese
Beschreibung des Konzepts: Die CSCHypothese beruht auf der bereits vor über hundert Jahren formulierten Annahme, dass eine spezielle Gruppe von Zellen mit Stammzell-Eigenschaften für die Entstehung, Progression und Persistenz eines Malignoms verantwortlich ist (Cohnheim, 1875).
Definitionsgemäß haben diese Zellen die Fähigkeit zur unbegrenzten Selbsterneuerung und Differenzierung, verhalten sich also in vielen Aspekten wie normale adulte Stammzellen. Diese Persistenz und das divergente Entwicklungspotenzial sind die Basis für die Genese verschiedenster Zelltypen einer Neoplasie und damit auch für die Heterogenität, welche besonders bei soliden Tumoren sehr ausgeprägt sein kann. Im Gegensatz zu den Tumorstammzellen können sich andere Zellen aus einem Tumor nicht mehr unbegrenzt selbst erneuern und auch nicht (mehr) in alle verschiedenen Zelltypen eines Tumors differenzieren. Man nimmt an, dass CSC aus normalen Stammzellen oder Vorstufenzellen hervorgehen und als kleine Population innerhalb eines Tumors persistieren. Bei der akuten myeloischen Leukämie ist es schon recht gut dokumentiert, dass es sich bei den Leukämie- Stammzellen (LSC) um Progenitorzellen handelt, die ein abnormales Potenzial zur ständigen Selbsterneuerung entwickelt haben (Goardon et al., 2011). Die Progression eines Tumors resultiert nach dieser Hypothese aus der metastatischen Ausbreitung dieser Zellen, und die Persistenz eines Tumors hängt mit deren Therapieresistenz zusammen (Greaves, 2010).
Unterstützende Beweise: Die Tumorstammzellhypothese wird von zahlreichen biologischen Beobachtungen unterstützt. Abbildung 1 zeigt in vereinfachter Weise die streng hierarchische Struktur des „klassischen“ Tumorstammzellmodells.
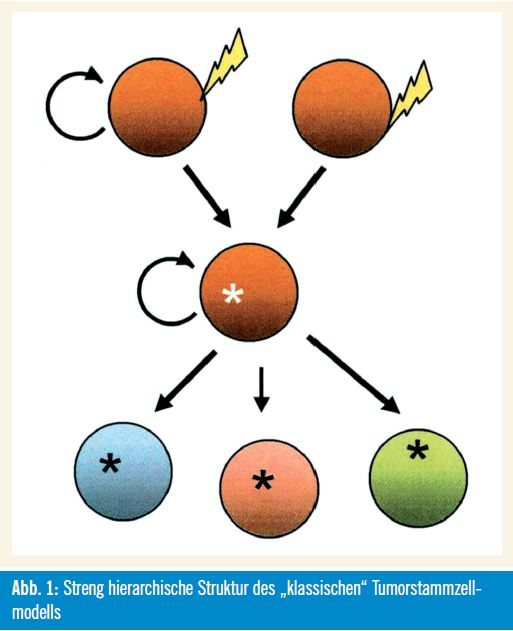
Die „klassische“ Charakteristik von Tumoren, welche die unbegrenzte Kapazität zur Selbsterneuerung und Proliferation und die phänotypische Heterogenität, die auch verschiedenste Differenzierungsstadien von nicht teilungsfähigen Zellen inkludiert, kann dadurch erklärt werden, dass Tumoren aus selbsterneuernden, multipotenten und langsam teilenden Zellen hervorgehen. Außerdem wird die Funktionalität normaler Stammzellen wie auch jener von Tumorstammzellen von epigenetischen Mechanismen und durch die Kommunikation mit der zellulären Umgebung (Microenvironment) reguliert. Beide Zelltypen haben viele Eigenschaften bzw. Fähigkeiten gemeinsam, wie die Induktion der Angiogenese, die Resistenz gegen Apoptose und Medikamente sowie die Zellmigration. Dies inkludiert auch für Tumorstammzellen typische Eigenschaften wie die Fähigkeit zur Induktion bzw. zum Nachwachsen sowie zur Metastasierung von Tumoren.
Das Modell der klonalen Evolution
Beschreibung des Konzepts: Das Modell der klonalen Evolution beruht daruf, dass Krebszellen verschiedenste Kombinationen von Mutationen erwerben, wobei sich die aggressivsten Zellen durchsetzen und die Progression des Tumors vorantreiben. Nach diesem Modell bietet eine Serie von Mutationen einer einzelnen malignen Zelle einen Selektionsvorteil gegenüber den angrenzenden normalen Zellen. Im weiteren Verlauf der Krebserkrankung nimmt die genetische Instabilität zu, es entstehen Zellen mit neuen Mutationen und Eigenschaften, die auch eine Resistenz gegen Apoptose implizieren können und zur Metastasierung bzw. zur Therapieresistenz führen.
Unterstützende Beweise: Nowell hat das Konzept der klonalen Evolution bereits 1976 entwickelt. Er hat postuliert, dass Tumoren im Lauf ihrer Progression entdifferenzieren, und interpretierte dies als Ursache für die Maximierung der Proliferation und der Invasivität. Dieser Effekt kann durch einen Selektionsdruck innerhalb eines Tumors erzielt werden. Die zunehmende genetische Instabilität der Tumorzellen wird in diesem Kontext als evolutionärer Vorteil gedeutet. Auch die Monoklonalität, die unbegrenzte Kapazität zur Proliferation und die Heterogenität der Zellen, die sich durch verschiedenste morphologische und metabolische Eigenschaften sowie durch ihre spezifische Kommunikation mit dem so genannten Microenvironment auszeichnen, wird in diesem Modell berücksichtigt.
Weiters passt die Entwicklung von therapieresistenten Klonen, wie sie z. B. nach Behandlung mit dem Tyrosinkinaseinhibitor Imatinib auftreten können, zu diesem Modell. Schließlich weisen Studien zu verschiedensten Tumoren auf genetische Ähnlichkeiten zwischen den Primärtumoren und Metastasen hin, die als das Ergebnis einer klonalen Evolution gedeutet werden könnten.
Wie in Abbildung 2 gezeigt wird, haben im klonalen Evolutionsmodell alle undifferenzierten Zellen die gleiche tumorigene Kapazität (Nowell, 1976). Mutierte Zellen mit einem Wachstumsvorteil werden selektiert und expandiert, wobei die Zellen der dominanten Population ein ähn liches Potenzial zur Re – generierung des Tumorwachstums aufweisen. Das (sequenzielle) Auftreten genetischer Veränderungen unterstützt dieses Modell.
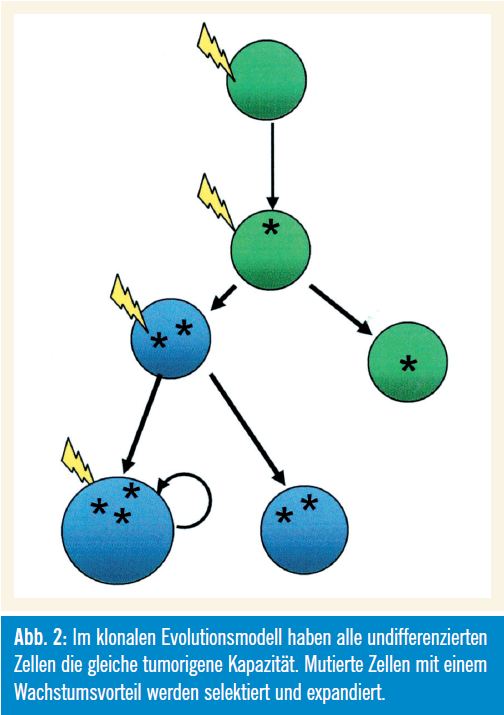
Grenzen des Modells: Interessanterweise zeigt gerade die von Nowell als klassisches Modell für die klonale Evolution herangezogene Philadelphia-Chromosom- positive CML auch die Grenzen dieses Modells auf. Die Inaktivierung der auf diesem Chromosom kodierten BCRABL- Tyrosinkinase mit Imatinib kann nämlich nicht die Leukämie-Stammzellen (LSC) eliminieren, die für einen Relaps der Erkrankung verantwortlich sind.
Vergleich der beiden Konzepte
Gemeinsamkeiten: In gewissen Aspekten stimmen die Stammzellhypothese und das Modell der klonalen Evolution überein, da nach beiden Modellen die Tumoren aus einzelnen Zellen hervorgehen, die eine Reihe von Mutationen und ein unbegrenztes Proliferationspotenzial aufweisen. In beiden Fällen wird die Exis tenz einer typischen Ursprungszelle („Cell of Origin“) postuliert. Weiters berücksichtigen beide Modelle die Bedeutung von Faktoren der unmittelbaren Umgebung („Microenvironment“), die das Aussehen eines Tumors und die klinischen Parameter bestimmen. Außerdem postulieren beide Modelle die Existenz von tumorigenen Zellen mit stammzelltypischen Eigenschaften, die einen selektiven Wachstumsvorteil gegenüber anderen (neoplastischen) Zellen aufweisen.
Unterschiede: Es gibt auch einige grundlegende Unterschiede zwischen der Stammzellhypothese und dem Modell der klonalen Evolution:
Zunächst erklären sie die Heterogenität von Tumoren mit verschiedenen Mechanismen: Die Tumorstammzellen zeichnen sich durch ein Programm zur aberranten Differenzierung aus, während beim klonalen Modell eine Vielfalt von Zelltypen durch kompetitive Mechanismen und Selektionsprozesse entsteht. Außerdem unterliegen nach der Stammzellhypothese vor allem die normalen Stamm- und Vorstufenzellen der malignen Transformation, während es im klonalen Evolutionsmodell eigentlich keine normalen Zellen gibt. Drittens beruht die Stammzellhypothese darauf, dass nur eine kleine Population von Zellen – eben die Tumorstammzellen – zur Progression eines Tumors beiträgt, während nach dem klonalen Evolutionsmodell jede Zelle in diesen Prozess involviert sein kann. Nur die Tumorstammzelle hat in diesem Modell das Potenzial, weiter zu mutieren und aggressiver zu werden – ein Vorgang, der nach dem klonalen Evolutionsmodell in jeder Tumorzelle stattfinden kann. Die Selbsterneuerungskapazität kann daher von unterschiedlichen Zellpopulationen erworben werden: entweder nur von den Tumorstammzellen oder von jeglichem Zelltyp.
Ein anderer Unterschied zwischen den beiden Theorien betrifft die Erklärungsmodelle für Therapieresistenz: Entweder sind nur die Tumorstammzellen therapieresistent oder die Therapie selektiert für entsprechend tolerante Klone.
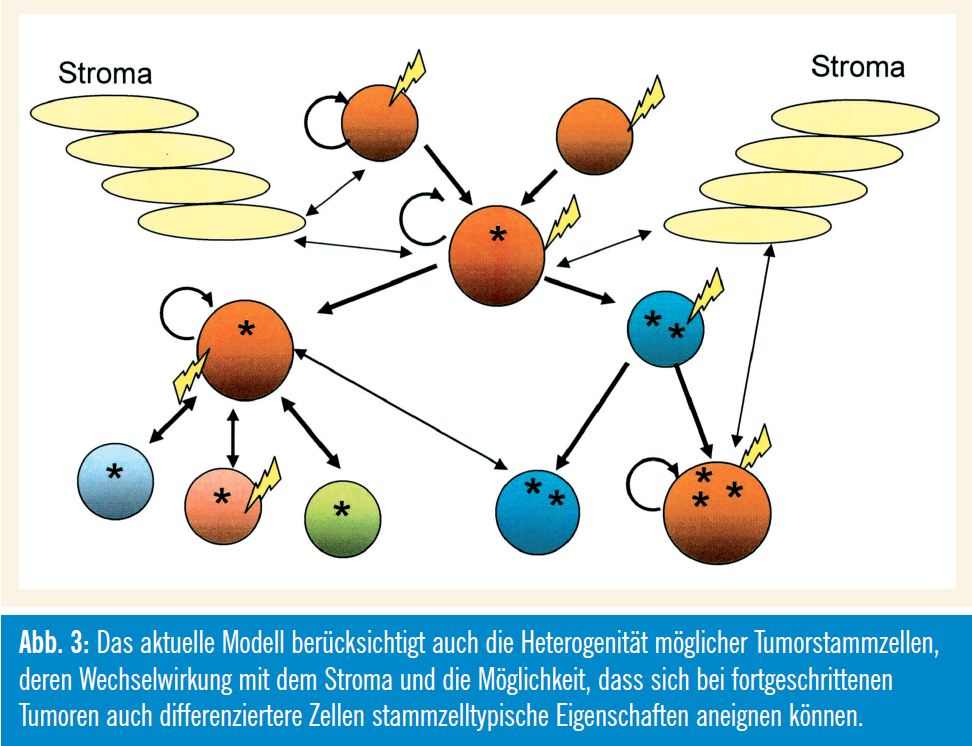
Welche Daten unterstützen das eine und das andere Modell der Krebsentstehung?
Beispielsweise unterstützen Ergebnisse aus der Brustkrebsforschung verschiedene Aspekte der Tumorstammzellhypothese. Erstens gibt es Hinweise darauf, dass Brustkrebs-Stammzellen (Tumor-CD44+- Zellen) aus normalen Stammzellen hervorgegangen sind, weil sie viele Stammzellmarker exprimieren, die auch auf normalen CD44+-Stammzellen vorkommen, welche üblicherweise gegen Ende einer Schwangerschaft verschwinden. Zweitens können Tumor-CD44+-Zellen der Ausgangspunkt für eine „Hierarchie“ von Tumorzellen sein: Die Tumor-CD24-Zellen sind den CD24-Zellen in der normalen Brustdrüse ähnlich und können genetische Veränderungen aufweisen, die man in Tumor-CD44+-Zellen nicht findet. Drittens haben die Tumor-CD44+- Zellen einen ausgeprägteren migratorischen, angiogenen und invasiven Phänotyp, was darauf hinweist, dass diese Tumorstammzellen ein höheres Potenzial zur Metastasierung haben.
Beispiele aus der Hämatologie zeigen, dass CSC nicht unbedingt durch Transformation normaler Stammzellen entstehen müssen, sondern auch aus Vorstufenzellen und differenzierteren Zellen hervorgehen können, die wieder eine Selbsterneuerungskapazität erlangt haben. Eine Konsequenz dieses Modells ergibt sich daher aus den mechanistischen Parallelen zwischen den Selbsterneuerungsprogrammen von normalen Stammzellen und CSC. In vielen Fällen wurde angenommen, dass der maligne Prozess in differenzierteren Zellen initiiert wird: Bei hämatologischen Neoplasien trifft das z. B. auf so genannte Mixed – Lineage-Leukämien zu, bei denen durch verschiedene Gen-Rearrangements stammzelltypische Eigenschaften in differenzierteren Vorstufenzellen induziert werden.
Probleme mit der Identifizierung von Tumorstammzellen
Die Verfahren zur Identifizierung von Tumorstammzellen aus soliden Tumoren werden nach wie vor kritisch diskutiert: Um Subpopulationen von Zellen zu isolieren, werden die Tumoren üblicher – weise mit proteolytischen Enzymen behandelt und dann nach Färbung mit verschiedenen Antikörpern und/oder Hoechst 33342 mittels Flowzytometrie oder mit immunomagnetischen Kügelchen sortiert. Diese Behandlung könnte einen gewissen Selektionsprozess darstellen.
Auch der Nachweis der Tumorigenität durch Injektion in immundefiziente Mäuse muss nicht unbedingt das Verhalten dieser Zellen im Patienten widerspiegeln, allein schon, wenn man die Unterschiede zwischen diesen beiden Spezies berücksichtigt, aber noch mehr, wenn man die Rolle der zellulären Umgebung (Microenvironment) in Betracht zieht, das in aktuellen Modellen immer mehr an Bedeutung gewinnt.
Das bedeutet, dass die identifizierten „Tumorstammzellen“ nicht die einzigen tumorigenen Zellen eines Menschen sein müssen und daher auch nur einen kleinen Prozentsatz der Tumorzellen darstellen.
Eine der wichtigsten Annahmen der Tumorstammzellforschung, nämlich dass nur eine kleine Population von Zellen kanzerogen sein muss, könnte daher unrichtig sein. Neuere Daten vom Glioblastom, aber auch vom Melanom (Weinberg et al., 2010) weisen darauf hin, dass ein relativ hoher Anteil der Tumorzellen Stammzellpotenzial haben kann, was dazu geführt hat, dass in aktuelleren Übersichtartikeln der Begriff der „Ursprungszellen“ (Cells of Origin) statt dem Stammzellbegriff verwendet wird (Visvader, 2011). Für die Verbesserung diverser Therapiekonzepte ist es daher besonders wichtig, die Biologie jener Neoplasien zu erforschen, bei denen die Eliminierung einer größeren Stammzellpopulation notwendig ist, wobei auch die zelluläre Umgebung (Microenvironment, Stroma) in Betracht gezogen werden muss (Karlic et al., 2010; Valent, 2011).
Die immer größer werdende Menge an epigenetischen Befunden liefert wichtige Indizien dafür, dass die Entstehung von Tumorstammzellen nicht unbedingt mit einer „Evolution“ zusammenhängen muss, sondern recht spontan passieren kann (Ehrlich, 2009). Als Ursprungszellen kommen hierbei sowohl unreife als auch differenziertere Zellen in Frage.
Indem man Methylierungsmuster von nichtexprimierten Genen verfolgte, konnte man nachweisen, wie schnell individuelle Stammzellen innerhalb von Krypten des Dickdarms dominant werden können. Mathematische Modelle zeigten, dass die Dynamik der Stammzellen und eine so genannte epigenetische Drift für die unterschiedlichen Methylierungsmus – ter verantwortlich war (Graham et al., 2011).
Schlussfolgerung
Nach dem derzeitigen Stand der Erkenntnisse müssen hierarchische bzw. klonale Aspekte des Stammzellmodells in holistischere und dynamischere Modelle integriert werden, bei denen auch die dynamische Umwandelbarkeit verschiedener Phänotypen (= Plastizität) berücksichtigt wird.
Zusammenfassung
Tumoren setzen sich aus morphologisch und physiologisch unterschiedlichen Zell – typen zusammen. Die Entstehung dieser Heterogenität wurde bislang vor allem mit zwei Konzepten erklärt: der Stammzellhypothese, die eine mehr oder weniger große Population von Tumorstammzellen als Wurzel der Entstehung und Persistenz von Malignomen postuliert, und dem älteren Modell der klonalen Evolution, bei dem sich die Neoplasie ausgehend von einer einzelnen Zelle durch Mutations- und Selektionsprozesse entwickelt. Wenn es auch Gemeinsamkeiten zwischen diesen Konzepten gibt, so bestehen doch fundamental verschiedene Ansätze mit unterschiedlichen klinischen Konsequenzen: Es ist sicher wichtig und notwendig, nach einer definierten Stammzelle zu fahnden und diese gezielt zu vernichten – aber reicht das wirklich aus, um zu verhindern, dass aus verbleibenden, differenzierteren Tumorzellen wieder Malignome entstehen?
Nach neueren Publikationen ist Letzteres nicht auszuschließen – es gibt sogar immer mehr Hinweise darauf, dass nach der Eradikation eines Tumors wieder weitere Malignome bzw. Metastasen aus Zellen entstehen können, die aufgrund ihrer phänotypischen „Normalität“ nicht als maligne Zellen erkannt und vernichtet werden können. Bei der Entwicklung effizienterer Ansätze zur Therapie und Prävention maligner Erkrankungen müssen daher komplexere Zusammenhänge in Betracht gezogen werden, die beide Konzepte in systemische Modelle integrieren.
FACT-BOX
• Tumorstammzellhypothese: Definierte Tumorstammzellen (CSC) mit unbegrenzter Selbsterneuerungsfähigkeit sind die Ursache für die Persistenz einer Neoplasie => die gezielte Vernichtung der CSC kann den Tumor vernichten.
• Modell der klonalen Evolution: Krebszellen erwerben verschiedenste Kombinationen von Mutationen => die aggressivsten Zellen setzen sich durch => möglichst alle Tumorzellen müssen durch die Therapie vernichtet werden.
• Neuere Erkenntnisse unterstützen kombinierte Modelle, inkludieren auch Aspekte der Umwandelbarkeit verschiedenster Zelltypen (Plastizität) und berücksichtigen die Bedeutung der Interaktion mit der zellulären Umgebung.
• Das Tumorstammzellmodell wird in systembiologische Modelle integriert.
Literatur:
– Cohnheim V (1875), Congenitales, quergestreiftes Muskelsarkom der Nieren. Virchows Archiv für Pathologische Anatomie und Physiologie und für Klinische Medizin 65:64–69
– Ehrlich M (2009), DNA hypomethylation in cancer cells. Epigenomics 1:239–259
– Goardon N et al. (2011), Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell 19:138–152
– Graham TA et al. (2011), Use of methylation patterns to determine expansion of stem cell clones in human colon tissue. Gastroenterology 140:1241–1250 e1241–1249
– Greaves M (2010), Cancer stem cells: back to Darwin? Semin Cancer Biol 20:65–70
– Karlic H, Herrmann H, Schulenburg A, Grunt TW et al. (2010); (Tumor stem cell research – basis and challenge for diagnosis and therapy. Wien Klin Wochenschr 122:423–436
– Nowell PC (1976), The clonal evolution of tumor cell populations. Science 194:23–28
– Valent P (2011), Targeting of leukemia-initiating cells to develop curative drug therapies: straightforward but nontrivial concept. Curr Cancer Drug Targets 11:56–71
– Visvader JE (2011), Cells of origin in cancer. Nature 469:314–322
– Weinberg R, Fisher DE, Rich J (2010), Dynamic and transient cancer stem cells nurture melanoma. Nat Med 16:758

Ursprünglich erschienen:
SO 05|2011
SO 05|2011
