
























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Tumorpromotor oder Tumorsuppressor? – Bedeutung der Sirtuine in der Prävention und Pathogenese von Krebserkrankungen
23. Dezember 2011
Die Familie der Sirtuinproteine
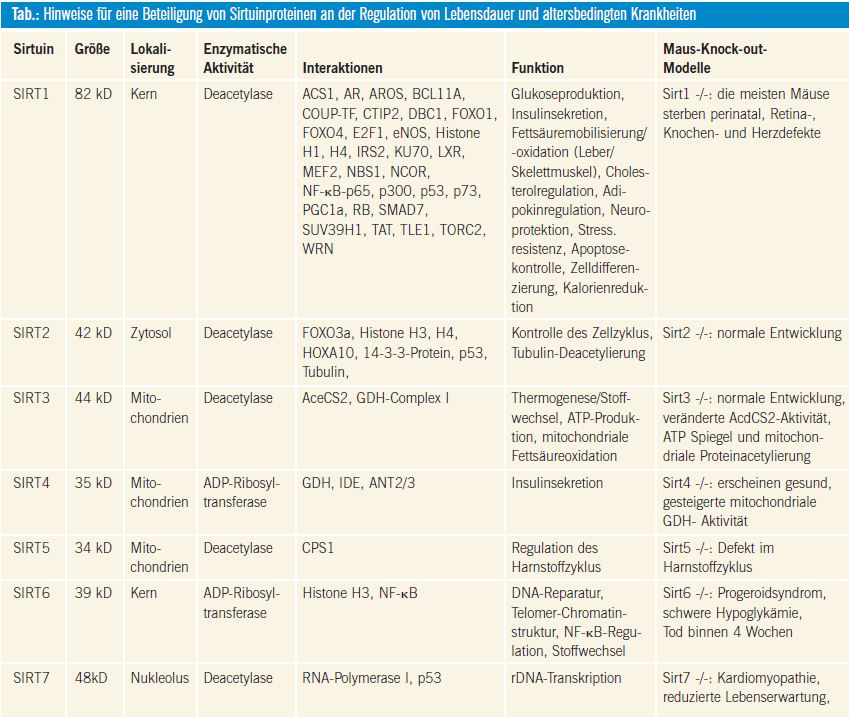 Bisher wurden sieben humane Sirtuine (SIRT1–SIRT7) identifiziert. Basierend auf phylogenetischen Analysen werden diese in vier Unterklassen eingeteilt1. Das Hauptmerkmal zur Unterscheidung der Sirtuine von den Nicht-Sirtuin-HDAC sind ihre einzigartigen enzymatischen Mechanismen. Die Sirtuine sind NAD+- abhängige Deacetylasen und ADP-Ribosyltransferasen. Die meisten Sirtuine katalysieren die NAD+-abhängige Deacetylierung. Während SIRT4 eine NAD+- abhängige Mono-ADP-Ribosyltransferase- Aktivität aufweist, haben SIRT1 und SIRT6 eine ADP-Ribosyltransferase-Aktivität und eine substratspezifische Deacetylase- Aktivität. Für SIRT4 und SIRT7 konnte eine Deacetylase-Aktivität bisher nicht nachgewiesen werden, möglicherweise ist eine solche enzymatische Aktivität nur in Anwesenheit eines spezifischen Substrats möglich, so wie z. B. bei SIRT6. Die enzymatischen Aktivitäten der Sirtuine sind mit vielen zellulären Prozessen assoziiert, wie z. B. mit dem Silencing von Heterochromatin, der Zelldifferenzierung, metabolischen Vorgängen, neuronalen Schutzmechanismen sowie mit zellulärer Apoptose und mit dem Überleben von Zellen, was auf der Fähigkeit der Sirtuine beruht, sowohl His tone als auch multiple Nicht-Histon- Ziele zu deacetylieren. Drei Säuger-Sirtuine konnten innerhalb der Mitochondrien (SIRT3, SIRT4 und SIRT5) lokalisiert werden, während die anderen Sirtuine ihre Funktionen innerhalb des Zytosols (SIRT2), im Zellkern (SIRT1 und SIRT6) oder innerhalb der Nukleolen (SIRT7) ausüben (Tab.). Eine Reihe von Signaltransduktionswegen ist mit der Fähigkeit einer Kalorienrestriktion assoziiert, die Lebensdauer zu verlängern. In diesem Zusammenhang spielen die Sirtuine eine zentrale Rolle, da diese NAD+ als Kofaktor für die enzymatische Aktivität benötigen, was auf ihre Bedeutung bei der energieabhängigen Regulation der Genexpression hinweist. In der Tat konnten Untersuchungen an niederen Organismen (Hefe, Drosophila melanogaster oder Caenorhabditis elegans) zeigen, dass sowohl eine Überexpression wie auch eine gesteigerte Aktivität von SIR2 (Hefe) und ihren Orthologen mit einer verlängerten Lebensdauer einhergeht (Tab., Abb. 1).
Bisher wurden sieben humane Sirtuine (SIRT1–SIRT7) identifiziert. Basierend auf phylogenetischen Analysen werden diese in vier Unterklassen eingeteilt1. Das Hauptmerkmal zur Unterscheidung der Sirtuine von den Nicht-Sirtuin-HDAC sind ihre einzigartigen enzymatischen Mechanismen. Die Sirtuine sind NAD+- abhängige Deacetylasen und ADP-Ribosyltransferasen. Die meisten Sirtuine katalysieren die NAD+-abhängige Deacetylierung. Während SIRT4 eine NAD+- abhängige Mono-ADP-Ribosyltransferase- Aktivität aufweist, haben SIRT1 und SIRT6 eine ADP-Ribosyltransferase-Aktivität und eine substratspezifische Deacetylase- Aktivität. Für SIRT4 und SIRT7 konnte eine Deacetylase-Aktivität bisher nicht nachgewiesen werden, möglicherweise ist eine solche enzymatische Aktivität nur in Anwesenheit eines spezifischen Substrats möglich, so wie z. B. bei SIRT6. Die enzymatischen Aktivitäten der Sirtuine sind mit vielen zellulären Prozessen assoziiert, wie z. B. mit dem Silencing von Heterochromatin, der Zelldifferenzierung, metabolischen Vorgängen, neuronalen Schutzmechanismen sowie mit zellulärer Apoptose und mit dem Überleben von Zellen, was auf der Fähigkeit der Sirtuine beruht, sowohl His tone als auch multiple Nicht-Histon- Ziele zu deacetylieren. Drei Säuger-Sirtuine konnten innerhalb der Mitochondrien (SIRT3, SIRT4 und SIRT5) lokalisiert werden, während die anderen Sirtuine ihre Funktionen innerhalb des Zytosols (SIRT2), im Zellkern (SIRT1 und SIRT6) oder innerhalb der Nukleolen (SIRT7) ausüben (Tab.). Eine Reihe von Signaltransduktionswegen ist mit der Fähigkeit einer Kalorienrestriktion assoziiert, die Lebensdauer zu verlängern. In diesem Zusammenhang spielen die Sirtuine eine zentrale Rolle, da diese NAD+ als Kofaktor für die enzymatische Aktivität benötigen, was auf ihre Bedeutung bei der energieabhängigen Regulation der Genexpression hinweist. In der Tat konnten Untersuchungen an niederen Organismen (Hefe, Drosophila melanogaster oder Caenorhabditis elegans) zeigen, dass sowohl eine Überexpression wie auch eine gesteigerte Aktivität von SIR2 (Hefe) und ihren Orthologen mit einer verlängerten Lebensdauer einhergeht (Tab., Abb. 1).
Die Bedeutung von Sirtuinen für die Karzinogenese
Obwohl eine Kalorienreduktion bei Nagetieren und Primaten scheinbar den effektivsten Weg der Krebsprävention darstellt – was von manchen Autoren als Hinweis auf die tumorsuppressive Wirkung von Sirtuinen gewertet wird –, scheinen einige Sirtuine, wie SIRT1 und SIRT3, das Überleben der Zelle zu verbessern, was für eine tumorfördernde Wirkung sprechen könnte. Steigende Krebsraten korrelieren direkt mit zunehmendem Alter. Die meisten Krebsarten entstehen aus genetischen oder epigenetischen Schäden im Bereich erneuerbarer Gewebsstrukturen, was mit gesteigerter Zellproliferation und Zellüberleben infolge beeinträchtigter Apoptose einhergehen kann. Während eine Degeneration von Organstrukturen in der Regel mit einem Funktionsverlust einhergeht, ist die Karzinogenese mit einer Zunahme von aberranten Funktionen assoziiert2, 3, 4.
Sirtuin-1, SIRT1
Der intrazelluläre SIRT1-Spiegel ist bei einer Reihe von Krebszellen deutlich erhöht. SIRT1 bindet und deacetyliert den Androgenrezeptor und unterdrückt den dihydrotestosteroninduzierten Androgen- Rezeptor-Signalweg in humanen Prostata- Krebszellen. Auf der anderen Seite induziert SIRT1 das Silencing von Genen wie auch Wachstum und Apoptose in menschlichen epithelialen Krebszellen. Die Induktion von SIRT1 in einem Beta- Catenin-abhängigen murinen Kolonkarzinom- Modell reduziert signifikant die Tumorbildung bzw. die Proliferation von Tumorzellen und damit die Morbidität der Tiere. Jedoch kann SIRT1 auch die TNF_-induzierte Apoptose stimulieren, was darauf hinweist, dass SIRT1 die Apoptose nicht nur unterdrücken, sondern auch fördern kann.
Neben der Deacetylierung von Histonen deacetyliert SIRT1 auch Nicht-Histon- Proteine, wie z. B. verschiedene Transkriptionsfaktoren, die an der Wachstumsregulierung, der Stressantwort und der Apoptose bei der Krebsprogression beteiligt sind. Die Hemmung von SIRT1 geht mit einem Anstieg der H4K16-, H3K9- und H1K26-Acetylierung bei endogenen Promotoren einher und reicht aus, um die Gen-Reexpression in Brustund Darmkrebs-Tumorzellen zu induzieren. SIRT1 reguliert auch die Bildung von Heterochromatin über eine Deacetylierung von Histon H1K26 und fördert den Verlust von H3K79me2, einem mit transkriptionell aktivem Chromatin assoziierten Marker. Dies sind nur einige Beispiele dafür, wie SIRT1 eventuell mit der Modulation epigenetischer Merkmale von Krebs assoziiert ist.
Beeinflussung von Tumorsuppressoren: Trotz aller positiven Effekte kann die SIRT1-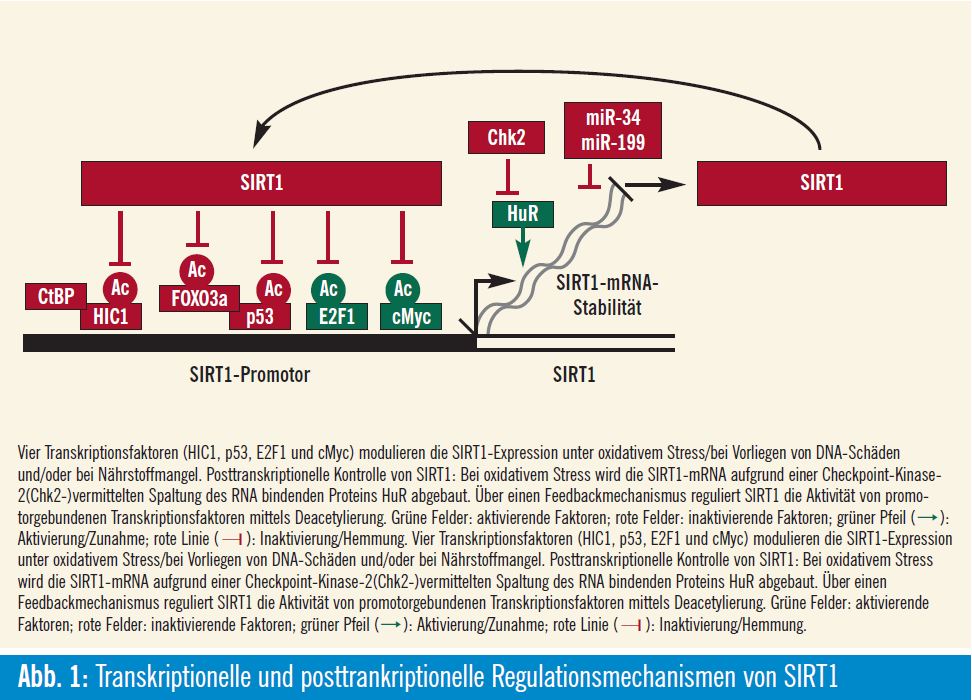 Aktivierung auch nachteilig sein: Die SIRT1-katalysierte Deacetylierung des Tumorsuppressorproteins p53 geht mit einer p53-Inaktivierung einher, was Zellen ermöglicht, die p53-vermittelte Apoptose (Abb. 1) zu umgehen. Das ist positiv für normale Zellen, da ihr Überleben gefördert und das Leben verlängert wird. In Tumorzellen ist dieser Effekt jedoch überhaupt nicht erwünscht, da er das Tumorwachstum verstärkt. Auf der anderen Seite reguliert eine Reihe von Tumorsuppressorproteinen, wie z. B. p53, HIC1 (hypermethylated in cancer 1) u
Aktivierung auch nachteilig sein: Die SIRT1-katalysierte Deacetylierung des Tumorsuppressorproteins p53 geht mit einer p53-Inaktivierung einher, was Zellen ermöglicht, die p53-vermittelte Apoptose (Abb. 1) zu umgehen. Das ist positiv für normale Zellen, da ihr Überleben gefördert und das Leben verlängert wird. In Tumorzellen ist dieser Effekt jedoch überhaupt nicht erwünscht, da er das Tumorwachstum verstärkt. Auf der anderen Seite reguliert eine Reihe von Tumorsuppressorproteinen, wie z. B. p53, HIC1 (hypermethylated in cancer 1) u
und DBC1, SIRT1 negativ: HIC1 ist eine Zinkfingerprotein, das durch p53 reguliert wird, was wiederum an den SIRT1- Promotor bindet und somit die Sirt1- Transkription unterdrückt. Dementsprechend reguliert die Inaktivierung von HIC1 die Transkription von SIRT1 hoch, wodurch p53 inaktiviert wird, wodurch Zellen die Apoptose nach DNA-Schädigung umgehen können. Interessanterweise scheint HIC1 eine vom Lebensalter abhängige Hypermethylierung in der Promotorregion zu erfahren, was zumindest teilweise die zunehmende Anfälligkeit für Krebs mit zunehmendem Lebensalter erklären könnte. CtBP ist ein Korepressor, der an HIC1 bindet, und diese Bindung von CtBP an HIC1 ist besonders stark im Rahmen der Glykolyse. Deshalb ist bei einer Hemmung der Glykolyse die Bindung des Korepressors CtBP an HIC1 verringert, was wiederum die SIRT1-Expression verstärkt (Abb. 1). DBC1 (deleted in breast cancer) ist ein weiterer Tumorsuppressor, der die SIRT1-Deacetylase-Aktivität negativ reguliert. Ein DBC1-Knock-down durch siRNA fördert die p53-Deacetylierung, wodurch wiederum Zellen genotoxischen Stress überleben können. DBC1 kann daher die Entwicklung von Brustkrebs durch die Aktivierung von SIRT1 fördern, was dann p53 und/oder andere Tumorsuppressorwege herunterreguliert. cMYC ist ein Protoonkogen, das die Zellproliferation, die Stammzellselbsterneuerung und die Apoptose reguliert. cMYC bindet an den SIRT1-Promotor und induziert die SIRT1-Expression, aber durch einen Feedbackmechanismus, der in der Tat eine zelluläre Transforma – tion vermeiden könnte, deacetyliert SIRT1 cMYC, was die cMYC-Stabilität reduziert und somit eine Tumor sup – pression induziert (Abb. 1)5, 6.
SIRT1: Tumorpromotor oder Tumorsuppressor? Tatsächlich scheint die SIRT1- Expression bei den meisten Krebsarten, wie z. B. beim Prostatakarzinom, bei der akuten myeloischen Leukämie, beim nichtmelanozytären Hautkrebs, beim primären Kolonkarzinom und beim Brustkrebs hochreguliert zu sein. Diese Tatsache und die Fähigkeit von SIRT1, eine Reihe von Proteinen, die mit Tumorsuppression und mit der Reparatur von DNA-Schäden assoziiert sind, zu inaktivieren, erklärt, warum SIRT1 vor allem als Tumorpromotor eingestuft wurde. Das SIRT1-vermittelte Silencing von ECadherin durch Hypermethylierung einer CpG-Insel auf der Promotor-Ebene ist ein Beispiel dafür, wie SIRT1 zur Karzinogenese in epithelialen Tumoren beitragen kann. Außerdem könnte die Reaktivierung von p53 durch eine SIRT1-Hemmung – anstelle einer Stimulation – die Apoptose von Tumorzellen triggern. Ob erhöhte intrazelluläre SIRT1-Spiegel Ursache oder Folge der Tumorbildung sind, ist derzeit noch ungeklärt. Im Gegenteil: in mehreren neueren Studien konnte gezeigt werden, dass die intrazellulären SIRT1-Spiegel in einigen anderen Krebsarten wie Glioblastomen, Harnblasen-, Prostata-, Mamma-, Ovarial- und Leberkarzinomen im Vergleich zu entsprechendem Normalgewebe reduziert waren und dass ein SIRT1-Mangel in der Tat zu genetischer Instabilität und Tumorgenese führen kann, während die Überexpression von SIRT1 die Krebsentstehung bei Mäusen, die heterozygot für den Tumorsuppressor p53 oder APC sind, dämpft, was auf der anderen Seite ein Hinweis dafür sein kann, dass SIRT1 eher ein Tumorsuppressor als ein Tumorpromotor in diesen Geweben ist. In Übereinstimmung mit aktuellen Publikationen ist SIRT1 wesentlich bei der Reparatur von DNA-Strangbrüchen und somit an der Prävention der Entwicklung zellulärer Malignität in murinen Karzinommodellen beteiligt, und in der Tat versterben Mäuse mit zusätzlichen SIRT1-Kopien nicht früher und entwikkeln nicht häufiger Krebs als die entsprechenden Kontrollmäuse. In murinen Leukämie- und Kolonkarzinommodellen wurde beobachtet, dass SIRT1-transgene Mäuse länger leben. Die Frage, ob SIRT1 in erster Linie als ein Onkogen oder als Tumorsuppressor wirkt, ist noch ungeklärt. Es ist jedoch unbestritten, dass SIRT1 ein entscheidender Regulator im Rahmen der Entstehung maligner Tumoren ist.
Des Weiteren spricht die Apoptoseregulierung beim Mammakarzinom durch SIRT1 für eine direkte positive Wirkung von SIRT1 bei Krebs. In gesunden Zellen wird die SIRT1-Expression durch BRCA1, einen potenten Tumorsuppressor, aufrechterhalten, was wiederum die Expression von Survivin, einem apoptosehemmenden Protein, inhibiert. Wenn BRCA1 durch eine Spontanmutation oder durch eine vererbte Mutation defekt ist, kann die defekte BRCA1 keine ausreichenden SIRT1-Spiegel mehr aufrechterhalten, und infolgedessen wird die Expression von Survivin möglicherweise nicht mehr ausreichend gehemmt, was zu einer Apoptoseresistenz und damit zu kontinuierlichem Tumorzellwachstum führt. Zumindest in vitro und in Tiermodellen konnte Resveratrol, das bekanntermaßen die SIRT1-Aktivität erhöht, das Tumorwachstum in BRCA1- defekten Zellen als Folge der reduzierten Survivin-Expression und der anschließenden Apoptose von BRCA1-defizienten Krebszellen stark hemmen.
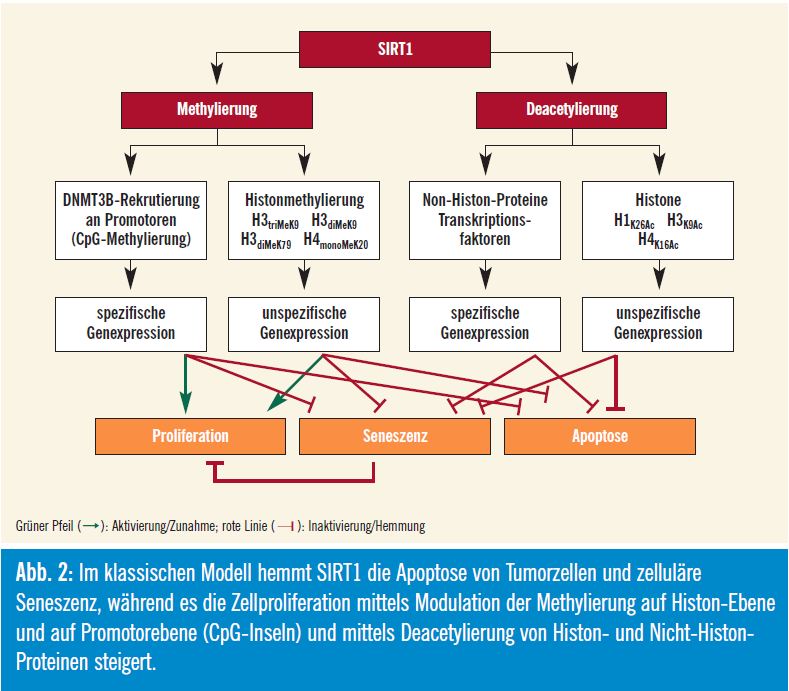
Neben seiner Aktivität als Deacetylase beeinflusst SIRT1 bekanntermaßen die Promotoren von etlichen fälschlicherweise supprimierten Tumorsuppressorgenen, deren DNA hypermethyliert ist. Dies ist besonders wichtig, wenn ein DNABruch innerhalb einer CpG-Insel initiiert wird, weil SIRT1 dann scheinbar für die vorübergehende Rekrutierung der DNAMethyltransferase 3B (DNMT3B) und die anschließende Suppression dieser DNA-Region durch Methylierung essenziell ist. Die Lokalisierung von SIRT1 im Bereich von DNA-Brüchen ist für eine effiziente Reparatur solcher DNA-Brüche notwendig: Zellen und Mäuse mit fehlendem SIRT1 sind anfälliger für durch DNA-Schäden induzierte Aneuploidie, und die Effizienz der Reparatur von DNABrüchen und die Aufrechterhaltung der Genomstabilität ist um 50 % reduziert. Zellen mit fehlendem SIRT1 sind nicht in der Lage, DNA-Reparaturproteine im Anschluss an DNA-Schäden effektiv zu rekrutieren (Abb. 2).
Sirtuin-3, SIRT3
SIRT3 spielt eine wichtige Rolle in der Karzinogenese: SIRT3 aktiviert die Expression von MnSOD (Magnesium-Superoxid- Dismutase) und der Katalase durch die Förderung der Translokation des zytosolischen FOXO3a in den Zellkern. FOXO3a beeinflusst die Expression des Antioxidans MnSOD, das mitochondriales Superoxid zu H2O2 degradiert und damit die zelluläre Transformation reguliert. Daher führt der Verlust oder eine altersbedingte Expressionsabnahme von SIRT3 zu einer erhöhten Phosphorylierung von FOXO3a, was einen Export aus dem Zellkern induziert und so die onkogene Transformation durch Verbesserung der mitochondrialen ROS fördert, die zu genetischer Instabilität und zur Stabilisierung des hypoxieinduzierbaren Faktors (HIF) führen kann. So fungiert SIRT3 als Tumorsuppressor, und es sind eher die SIRT3-Funktionen im Zellkern bzw. im Zytosol als in den Mitochondrien, die seine Rolle bei der Regulation der antioxidativen Aktivität und zelluläre Transformation vermitteln, und es sollte daher überprüft werden, ob eine Verwendung von NAD+ die Zellen möglicherweise in die umgekehrte Richtung entlang der Pfade der Tumortransformation lenken kann, in denen SIRT3 entweder fehlt oder reduziert ist. Da erhöhte Mengen an SIRT3- mRNA mit Brust- und Schilddrüsenkrebs in Verbindung gebracht werden, ist es derzeit unklar, in welchem Ausmaß SIRT3 eher als Tumorsuppressor und nicht als Tumorpromotor wirkt.
Schlussfolgerungen und Perspektiven
In erster Linie stellen Sirtuine Suppressoren für einige Krebsarten dar, während sie unter anderen Umständen die Karzinogenese zu fördern scheinen. Es ist derzeit unklar, in welchem Umfang und unter welchen besonderen Umständen Sirtuin- Aktivatoren und/oder -Inhibitoren ihren Platz in der Therapie von altersbedingten Erkrankungen und Krebs finden werden. Während die SIRT1-Expression und -Aktivität unter nichtmalignen Bedingungen durch Tumorsuppressoren unterdrückt wird, kann die SIRT1-Expression und -Aktivität bei vorliegender Überexpression von Onkogenen oder bei reduzierter Aktivität von Tumorsuppressorproteinen erhöht sein, was letztendlich die Seneszenz und Apoptose blo – ckieren kann. Jedoch kann es auch die Angiogenese induzieren, Zellwachstum stimulieren und mit Chemotherapieresistenz einhergehen. Weitere Untersuchungen hinsichtlich des aufeinander abgestimmten Zusammenspiels der verschiedenen Sirtuine werden daher nicht nur zu einem detaillierteren Verständnis von Alterungsprozessen beitragen, sondern könnten auch zur Entwicklung neuer Strategien in der Therapie von Karzinomen und anderen altersbedingten Erkrankungen führen.
Danksagung
Das vorliegende Manuskript stellt eine gekürzte Version des in englischer Sprache erschienenen Artikels Voelter-Mahlknecht S. and Mahlknecht U., The sirtuins in the pathogenesis of cancer. Clinical Epigenetics 2010; 1(3-4):71–83, dar. Für die freundliche Genehmigung von Springer Science und Business Media bedanken wir uns.
1 Voelter-Mahlknecht S, Mahlknecht U. Cloning, chromosomal characteriza – tion and mapping of the NAD-dependent histone deacetylases gene sirtuin 1. Int J Mol Med 2006; 17:59–67
2 Mahlknecht U, Hoelzer D, Histone acetylation modifiers in the pathogenesis of malignant disease. Mol Med 2000; 6:623–44
3 Mahlknecht U, Ottmann OG, Hoelzer D, When the band begins to play: histone acetylation caught in the crossfire of gene control. Mol Carcinog 2000; 27:268–71
4 Mei S, Ho AD, Mahlknecht U. Role of histone deacetylase inhibitors in the treatment of cancer (Review). Int J Oncol 2004; 25:1509–19
5 Zschoernig B, Mahlknecht U, SIRTUIN 1: regulating the regulator. Biochem Biophys Res Commun 2008; 376:251–5
6 Zschoernig B, Mahlknecht U, Carboxy-terminal phosphorylation of SIRT1 by protein kinase CK2. Biochem Biophys Res Commun 2009; 381:372–7

Ursprünglich erschienen:
SO 05|2011
SO 05|2011
