





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
„Emerging Biomarker“ für Immun-Checkpoint-Inhibitoren: Bestimmung der Tumormutationslast
20. September 2018
Die Tumormutationslast (TMB) gewinnt als „Emerging Biomarker“ zunehmend an Bedeutung. In einer rezent publizierten Phase-III-Studie zeigte sich, dass die TMB unabhängig von der PD-L1-Expression ein Prädiktor für das progressionsfreie Überleben von NSCLC-Patienten unter Therapie mit Nivolumab ist (Matthew Hellmann et al., New Engl J Med 2018). In klinischen Studien mit Atezolizumab bei NSCLC oder dem Urothelkarzinom der Blase wurden ebenfalls TMB-Bestimmungen durchgeführt.
Die Beobachtung ist, dass bei hoher Mutationslast die Wahrscheinlichkeit für die Wirksamkeit der Immuntherapie steigt. Grundlage ist, dass Mutationen immunogen sind und eine tumorspezifische Immunantwort auslösen können. Tumormutationslast ist definiert als die Anzahl somatischer Mutationen in einem Tumor. Nicht-synonyme Mutationen, die zu Veränderungen in den Aminosäuren eines Proteins führen, können als Neoantigene eine Immunantwort hervorrufen (Campesato et al., Oncotarget 2015).
In einer rezenten Arbeit wurde die Tumormutationslast mit Mikrosatelliteninstabilität, dem MSI-Status, korreliert (Grasso et al., Cancer Discovery 2018). Mikrosatelliteninstabilität als Folge defekter DNA-Reparaturproteine geht mit einer hohen Mutationsrate und einem vermehrten Auftreten von Neoantigenen einher und wurde bereits (unabhängig vom Tumortyp) zur Indikation für die Immuntherapie mit Pembrolizumab. Es zeigte sich, dass MSI-high CRC auch eine hohe TMB hat und damit eine Subklasse der TMB-high-Tumoren darstellt. Durch die häufigen Mutationen in TMB-high-Tumoren werden in den Tumorzellen Gene mutiert, die zu Tumorwachstum, Umstellung des Zellmetabolismus und in weiterer Folge zur Unterdrückung der Immunantwort des Körpers führen.
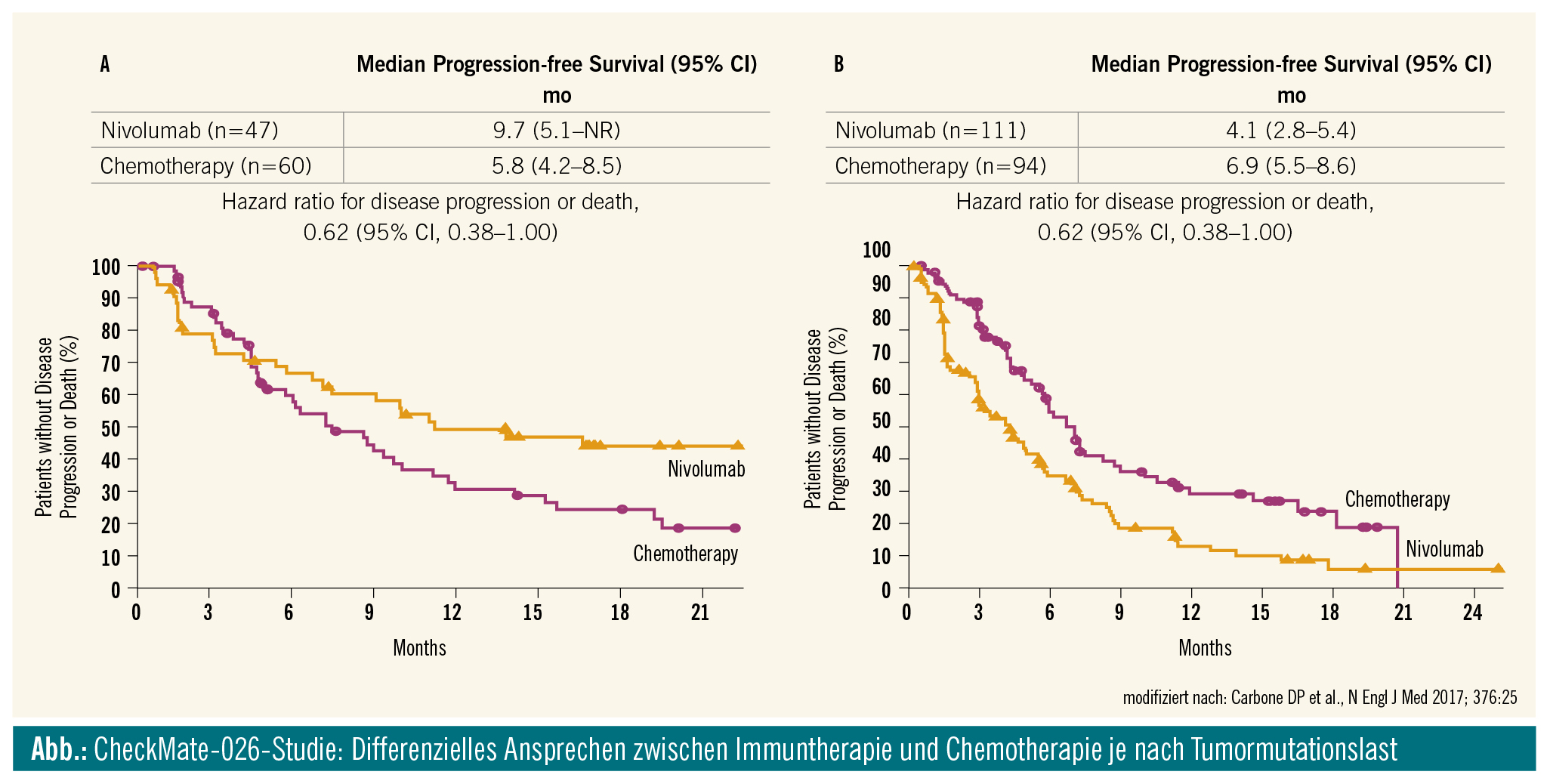
Im Kontext eines prädiktiven Markers für Immuntherapie wurde durch retrospektive Analyse eine Anzahl von mehr als 10 Mutationen pro Megabase als Cut-off für eine hohe TMB definiert (CheckMate-227-Studie, TMB-Testung FoundationOne). Klinisch ist dieser Cut-off sinnvoll, wie sich am unterschiedlichen Ansprechen zeigt. Allerdings muss man, um eine statistische Korrelation der ermittelten TMB mit dem Ansprechen auf Immuntherapie zu erreichen, eine relativ große Anzahl von Genen und damit Megabasen Tumorgenom sequenzieren. Als Goldstandard gilt das Whole Exome Sequencing, mit dem 44 Megabasen (23.000 Gene) abgegriffen werden, die Methode ist aber für die klinische Routine zu aufwendig. Für die Klinik zeichnet sich ab, dass Genpanels mit zumindest 1 Megabase analysiertem Tumorgenom in der Lage sind, eine aussagekräftige TMB anzuzeigen.
FoundationOne etwa untersucht 370 Gene (1,2 Megabasen). Pitfalls für die Vergleichbarkeit unterschiedlicher TMB-Bestimmungsmethoden bestehen u. a. darin, dass verschiedene Plattformen jeweils andere Gene untersuchen und für die Unterdrückung von Keimbahnvariationen unterschiedliche Datenbanken verwendet werden. Prinzipiell aber wäre es laut Dr. Kashofer möglich, mit den in der Pathologie zur Verfügung stehenden großen Genpanels im Rahmen der normalen Mutationsanalytik eine TMB herauszurechnen. „Sobald 300–400 Gene untersucht werden, sollten sich Tumoren mit hoher und niedriger Mutationslast unterscheiden lassen und die Ergebnisse vergleichbar sein.“

Ursprünglich erschienen:
Patho 01|2018
Patho 01|2018
