





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Periphere T-Zell-Lymphome
20. September 2018
Die angioimmunoblastischen T-Zell-Lymphome (AITL), die follikulären T-Zell-Lymphome (FTCL) und die nodalen peripheren T-Zell-Lymphome (nPTCL) mit follikulärem T-Helfer-Phänotyp zählen zu den peripheren T-Zell-Lymphomen (PTCL), die sich von der follikulären T-Helferzelle (TFH) ableiten. Es sind dies Lymphome mit charakteristischer Morphologie und besonderen klinischen und biologischen Merkmalen, wahrscheinlich hervorgerufen durch Funktionalität der neoplastischen TFH-Zellen mit Freisetzung von Zytokinen und Chemokinen sowie einer starken Wechselbeziehung zwischen Tumorzellen und Mikroenvironment.
Angioimmunoblastisches T-Zell-Lymphom
Die AITL stellen mit 15–30 % aller nicht-kutanen T-Zell-Lymphome bzw. 1–2 % aller Non-Hodgkin-Lymphome die größte spezifische Subgruppe der PTCL dar. Sie betreffen vorwiegend ältere Patienten, häufiger Männer. Es sind aggressive Lymphome mit einem medianen Überleben von unter drei Jahren. Sie werden meist in einem fortgeschrittenen Krankheitsstadium diagnostiziert. Klinisch zeigen betroffene Patienten neben einer generalisierten Lymphadenopathie häufig eine Hepatosplenomegalie sowie immunologische Symptome mit Hauteffloreszenzen, Ergüssen, Arthritiden, zirkulierenden Immunkomplexen, Kälteagglutininen mit hämolytischer Anämie und Rheumafaktoren. Eine polyklonale Hypergammaglobulinämie ist häufig nachweisbar.
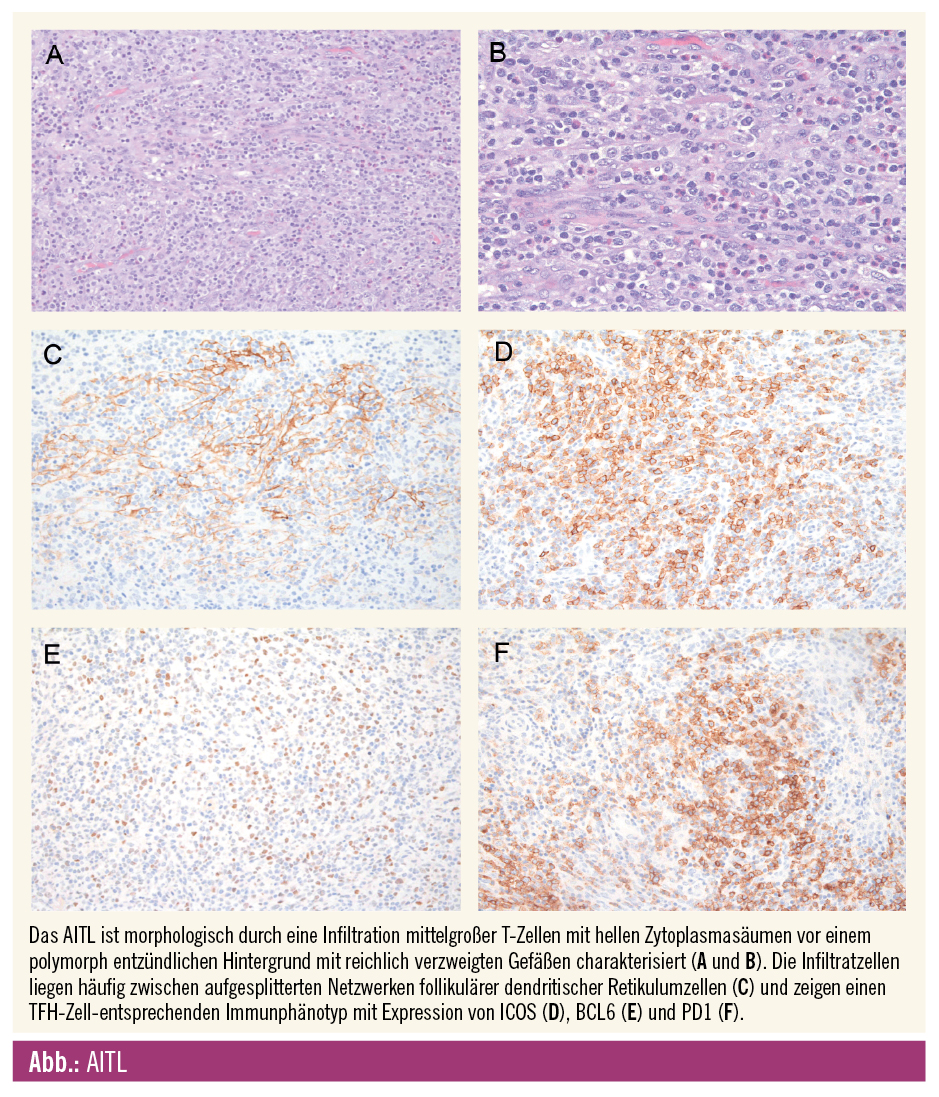
Histomorphologisch sieht man eine Infiltration aus kleinen bis mittelgroßen T-Zellen mit hellen, mittelbreiten Zytoplasmasäumen, deutlicher Zellmembran und geringer zellulärer Atypie vor einem variabel zusammengesetzten entzündlichen polymorphen Hintergrund aus Histiozyten, Plasmazellen und eosinophilen Granulozyten. Die Infiltratzellen liegen charakteristischerweise zwischen proliferierten verzweigten Venolen mit hohem Endothel und prominenten aufgesplitterten follikulären dendritischen Retikulumzellen (FD-Zellen). Oft sieht man eine Infiltration des perinodalen Weichgewebes mit Aussparung des Randsinus. Charakteristischerweise sieht man im Parakortex in variabler Dichte Epstein-Barr-Virus (EBV-)positive B-Immunoblasten. Auch Reed-Sternberg-artige Zellen (häufig EBV-positiv) können vorkommen und hier ein klassisches Hodgkin-Lymphom vortäuschen. EBV-positive B-Immunoblasten können im Verlauf, wahrscheinlich getriggert durch Zytokine der neoplastischen TFH-Zellen, auch sehr prominent werden und sogar zu einem EBV-positiven, diffus großzelligen B-Zell-Lymphom progredieren.
Je nach Ausmaß der neoplastischen Infiltration unterscheidet man 3 histomorphologische Wachstumsmuster: Pattern 1 ist durch eine nur sehr partielle und parakortikale neoplastische Infiltration um hyperplastische Keimzentren charakterisiert. Die Schwierigkeit liegt hier in der Abgrenzung zur follikulären Hyperplasie. Pattern 2 zeigt eine ausgedehntere parakortikale Infiltration um oftmals regressive Keimzentren. Pattern 3 ist durch eine gänzlich aufgehobene Lymphknotenarchitektur definiert. Mitunter können AITL einen sehr epitheloidzellreichen Hintergrund aufweisen, differenzialdiagnostisch müssen hier granulomatös-entzündliche Erkrankungen, andere histiozytenreiche B- und T-Zell-Lymphome (Lennert’s Lymphom) und das klassische Hodgkin-Lymphom ausgeschlossen werden.
Immunphänotypisch exprimieren die neoplastischen T-Zellen neben den Pan-T-Antigenen (CD2, CD3, CD5) überwiegend CD4 sowie TFH-Zellmarker wie CD10, ICOS, BCL6, PD1 und CXCL13 in einem variablen Ausmaß. CXCL13 und CD10 zeigen hierbei die höchste Spezifität, PD1 und ICOS die höchste Sensitivität. CXCL13 gilt als starker molekularer Mediator mit Einfluss auf die Keimzentrums-B-Zell-Rekrutierung und -B-Zell-Aktivierung und könnte somit die B-Zell-Expansion, plasmazytäre Differenzierung und Hypergammaglobulinämie fördern. Reaktive CD8-positive T-Zellen sind häufig untermengt.
Genetisch kann in 70–90 % der AITL-Fälle ein klonales T-Zell-Rezeptor-Gen-Rearrangement nachgewiesen werden, in 25–30 % der Fälle sogar auch ein klonales Immunglobulin-Gen-Rearrangement, zurückzuführen auf die Expansion von EBV-positiven B-Blasten.
Genexpressionsanalysen spiegeln die Bedeutung des Tumormikroenvironments wider: Die AITL zeigen hierbei eine eindeutige, mit einer Abstammung von der TFH-Zelle im Einklang stehende Gensignatur mit hoher Expression von B- und FD-Zellen-assoziierten Genen, Chemokinen, Chemokinrezeptoren und Genen mit Verwandtschaft zur extrazellulären Matrix und Gefäßbiologie. In einer großen multizentrischen Studie korrelierte eine Gensignatur mit hoher Expression von B-Zell-assoziierten Genen und einer niedrigen Expression monozytärer Gene mit einem verlängerten Überleben.
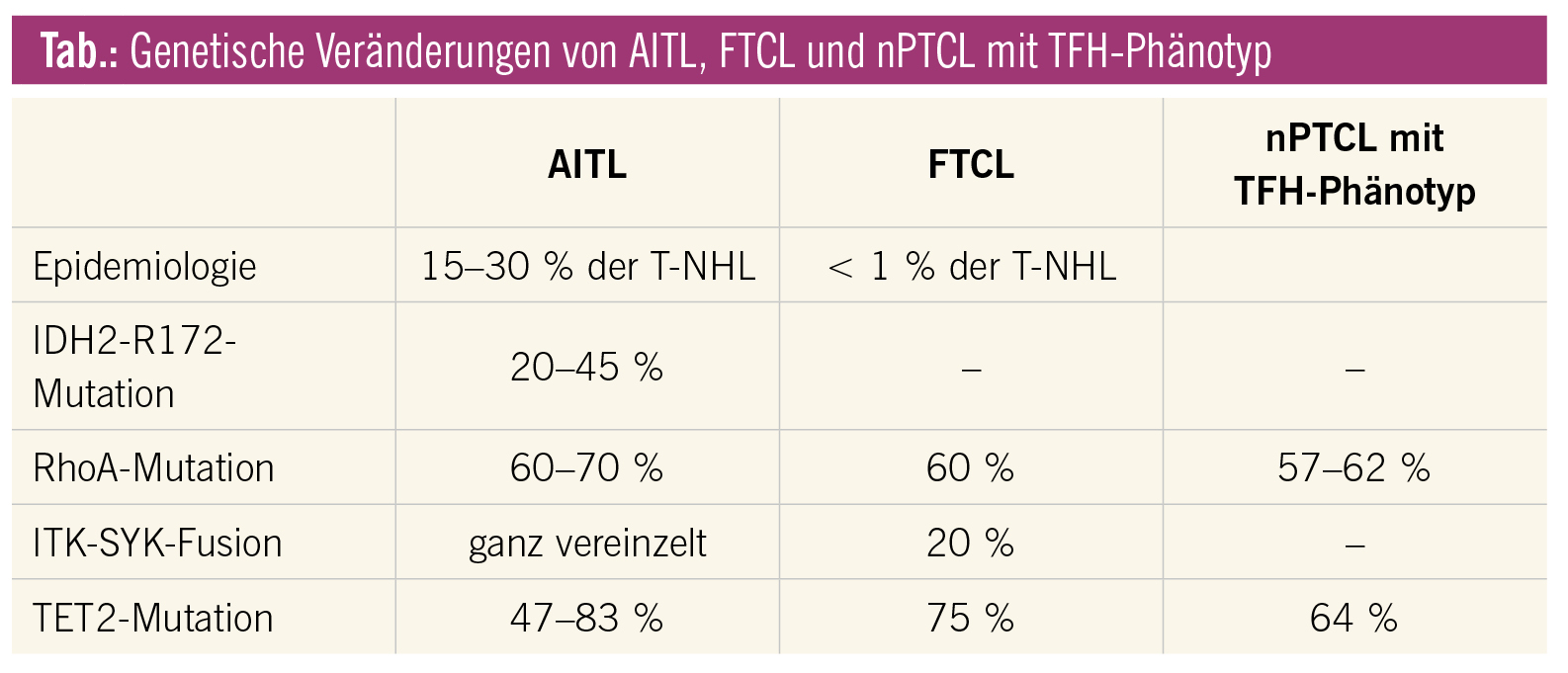
Zytogenetische Veränderungen sind Trisomien der Chromosomen 3, 5, 21, Zugewinn des Chromosoms X und Verlust von 6q. Auf DNA-Ebene ist in 60–70 % eine inaktivierende Mutation der kleinen GTPase RhoA (p.Gly17Val) nachweisbar. Des Weiteren finden sich Mutationen in den epigenetischen Modifier-Genen wie IDH2, TET2 und DNMT3A (Tab.). Während IDH-Mutationen auch in anderen Neoplasien, inklusive akute myeloische Leukämie (AML), vorkommen, findet man sie innerhalb der T-Zell-Lymphome ausschließlich in AITL und hier ausschließlich an der Position R172. Gene, die im T-Zell-Rezeptor-Signalweg eine Rolle spielen, wie FYN, PLCG1 und CD28, sind in 5–10 % mutiert. Mehr als 50 % der AITL zeigen eine CTLA4-CD28-Genfusion. Auch die eigentlich für die FTCL charakteristische t(5;9)(q33;q22) ITK-SYK-Genfusion ist selten auch in AITL detektierbar.
Follikuläres T-Zell-Lymphom
FTCL wurden früher den PTCL-NOS zugeordnet. Sie zeigen ein charakteristisches morphologisches und immunphänotypisches Bild und wurden erstmals 2001 von Leval beschrieben als „PTCL mit follikulärer Beteiligung und CD4+/BCL6+ Phänotyp“. Es sind dies sehr seltene Lymphome mit einem eher aggressiven klinischen Verlauf und starker Assoziation zum Keimzentrumsumfeld. Sie machen weniger als 1 % aller T-Zell-Lymphome aus und kommen vorwiegend in mittelalten bis älteren, häufiger männlichen Patienten vor. Diese Lymphome betreffen die Lymphknoten, gelegentlich kommt es auch zu einer Beteiligung von Haut und Knochenmark.
Die klinische Präsentation ähnelt jener der AITL mit üblicherweise fortgeschrittenem Krankheitsstadium, generalisierter Lymphadenopathie, Splenomegalie, B-Symptomen und Hautausschlägen. Auch AITL-typische Blutveränderungen wie Hypergammaglobulinämie, Eosinophilie oder ein positiver Coombs-Test können vorkommen.
Histomorphologisch zeigen FTCL eine partielle oder komplette Lymphknoteninfiltration mit einer nodulären/follikulären Proliferation mittelgroßer monomorpher lymphoider Zellen mit rundlichen Kernen und breiten, hellen Zytoplasmasäumen.
Zwei Wachstumsmuster können unterschieden werden: Das „follikuläre Lymphom-artige“ Wachstumsmuster ist durch eine Proliferation scharf umschriebener Knoten um eine ausgedünnte Mantelzone charakterisiert. Das „progressiv transformierte Keimzentren-artige“ Wachstumsmuster zeigt große, teils reguläre, teils irregulär begrenzte Knoten, aufgebaut aus kleinen Aggregaten neoplastischer T-Zellen, umgeben von IgD-positiven B-Mantelzonen-Zellen. Ohne Immunhistochemie kann eine Abgrenzung zu reaktiven Lymphknotenveränderungen und einigen B-Zell-Lymphomen (follikuläres Lymphom, Marginalzonenlymphom, noduläres lymphozytenprädominantes Hodgkin-Lymphom) schwierig sein. Einzelne Immunoblasten und Hodgkin-/Reed-Sternberg-artige Zellen können vorkommen, die für AITL typischen polymorphen Infiltrate und vaskulären Proliferate fehlen.
Immunphänotypisch exprimieren die neoplastischen Zellen Pan-T-Zellantigene (CD2, CD3 und CD5) mit häufiger Defizienz von CD7. Sie zeigen einen CD4+ TFH-Phänotyp mit Expression von zahlreichen TFH-Zellmarkern wie PD1, CXCL13, BCL6, CD10 und ICOS. Eingestreut liegende CD20-positive Immunoblasten sind häufig EBV-positiv. Hodgkin-/Reed-Sternberg-artige Zellen können auch den Phänotyp eines klassischen Hodgkin-Lymphoms (CD30+, CD15+, PAX5+, EBV+/-) aufweisen, wodurch das lymphozytenreiche klassische Hodgkin-Lymphom hier eine wichtige Differenzialdiagnose darstellt.
Die t(5;9)(q33;q22) ITK-SYK-Translokation wird als spezifische genetische Aberration angesehen und ist in etwa 20 % aller FTCL nachweisbar. FTCL zeigen ein starkes Naheverhältnis zu den AITL. Dies wird auch durch einige Fallberichte untermauert, wo die primäre Diagnose von FTCL im Krankheitsverlauf in einer späteren Biopsie zu einem typischen AITL und vice versa wechselte.
Nodales peripheres T-Zell-Lymphom mit TFH-Phänotyp
Periphere T-Zell-Lymphome mit einem TFH-Phänotyp wurden früher der großen Kategorie der PTCL-NOS zugerechnet und stellen nun in der rezenten WHO-Klassifikation eine eigene provisorische Subgruppe dar. Sie werden als nodales peripheres T-Zell-Lymphom mit follikulärem T-Helfer-Phänotyp bezeichnet. Sie sind CD4-positiv und müssen zumindest 2 (besser 3) der TFH-Marker (PD1, CD10, BCL6, CXCL13, ICOS) exprimieren. Oft zeigen sie morphologische Überlappungen zu den AITL, in der Regel sieht man jedoch dichte, diffuse Infiltrate ohne prominenten polymorphen Hintergrund und ohne vaskuläre oder FD-Proliferate. Genetisch zeigen diese Lymphome teilweise ebenso Überschneidungen zu den AITL – so finden sich auch hier TET2-, DNMT3A- und RhoA-Mutationen. Dies legt die biologische Verwandtschaft zu den AITL nahe und es wird diskutiert, ob diese Lymphome eventuell eine tumorzellreiche Variante der AITL darstellen könnten.
Resümee
T-Zell-Lymphome, die sich von der TFH-Zelle ableiten, zeigen charakteristische morphologische, klinische und genetische Veränderungen mit zahlreichen Überschneidungen innerhalb der Subgruppen. Sie sind charakterisiert durch einen TFH-Zell-Phänotyp mit variabler Expression von CD10, CXCL13, BCL6, ICOS, PD1 und starker Wechselbeziehung zum Mikroenvironment. Während die AITL die am besten charakterisierten Lymphome innerhalb dieser Gruppe sind, werden die seltenen FTCL und die nodalen PTCL mit TFH-Phänotyp in der neuen WHO-Klassifikation als provisorische Entität geführt. Es gilt hier noch zu klären, ob diese tatsächlich eine eigenständige Lymphomentität darstellen oder nur Teile des Spektrums der AITL sind, welches eventuell größer ist als bisher angenommen.

AutorIn: Priv.-Doz. Dr. Ana Iris Schiefer
Klinisches Institut für Pathologie,Medizinische Universität Wien, AKH Wien

Ursprünglich erschienen:
Patho 01|2018
Patho 01|2018
