





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Periphere T-Zell-Lymphome, not otherwise specified
20. September 2018
Das periphere T-Zell-Lymphom, not otherwise specified (PTL-NOS), repräsentiert eine heterogene Kategorie von nodalen und extranodalen, reifen T-Zell-Lymphomen, die sich nicht in anderen von der WHO definierten peripheren T-Zell-Lymphomentitäten wiederfinden.1 Neben dem angioimmunoblastischen T-Zell-Lymphom (AITL) ist das PTL-NOS, wie in der Tabelle angeführt, in Frankreich und wahrscheinlich auch bei uns das zweithäufigste nicht-kutane periphere T-Zell-Lymphom, bezogen auf die Gesamtheit der Non-Hodgkin-Lymphome aber trotzdem eine Rarität.2 Schon die Tatsache, dass es sich hier um einen „Sammeltopf“ für diagnostisch woanders nicht unterzubringende aggressive Lymphome handelt, unterstreicht den wissenschaftlichen Nachholbedarf.
Über die wichtigsten neuen pathogenetischen Erkenntnisse wird in diesem Artikel berichtet.

Klinische Merkmale des peripheren T-Zell-Lymphoms, NOS
Die meisten Patienten sind im mittleren und fortgeschrittenen Erwachsenenalter und präsentieren sich mit generalisierter Lymphadenopathie, mitunter auch mit primärer oder sekundärer Beteiligung von Haut und Gastrointestinaltrakt. Die extranodale Manifestation kann erhebliche differenzialdiagnostische Probleme hinsichtlich der Abgrenzung zu spezifischen kutanen und gastrointestinalen Entitäten bedeuten. B-Symptome (Gewichtsverlust, Fieber, Nachtschweiß) sind häufig.
Histologie und Immunphänotyp
Das zytologische Spektrum ist sehr breit, wobei mittelgroße und große pleomorphe Zellen dominieren. Ein entzündlicher Hintergrund ist oft vorhanden mit eosinophilen Granulozyten, Plasmazellen und histiozytären Zellen. Die meisten PTL-NOS exprimieren den α/β-T-Zell-Rezeptor, sind CD3+CD4+ mit variablem Verlust von CD5 und CD7. Häufig ist ein Teil der Zellen CD30+. Eine kleine Gruppe von PTL-NOS zeigt einen zytotoxischen Immunphänotyp (TIA1+, Granzyme B+), noch seltener besteht eine EBV-Assoziation.
Differenzialdiagnose
Die Abgrenzung gegenüber nicht-neoplastischen, reaktiven Prozessen kann im Einzelfall Probleme bereiten. Als Referenzbefunder bekommt man ein paar Mal pro Jahr einen von fleckförmigen Nekrosen durchsetzen Lymphknoten zugesandt, mit der Frage nach „partieller Infiltration durch ein peripheres T-Zell-Lymphom“, der sich als Kikuchi-Lymphadenitis herausstellt – ein ätiologisch unklarer entzündlicher Prozess, der zumeist nach einigen Wochen spontan ausheilt.
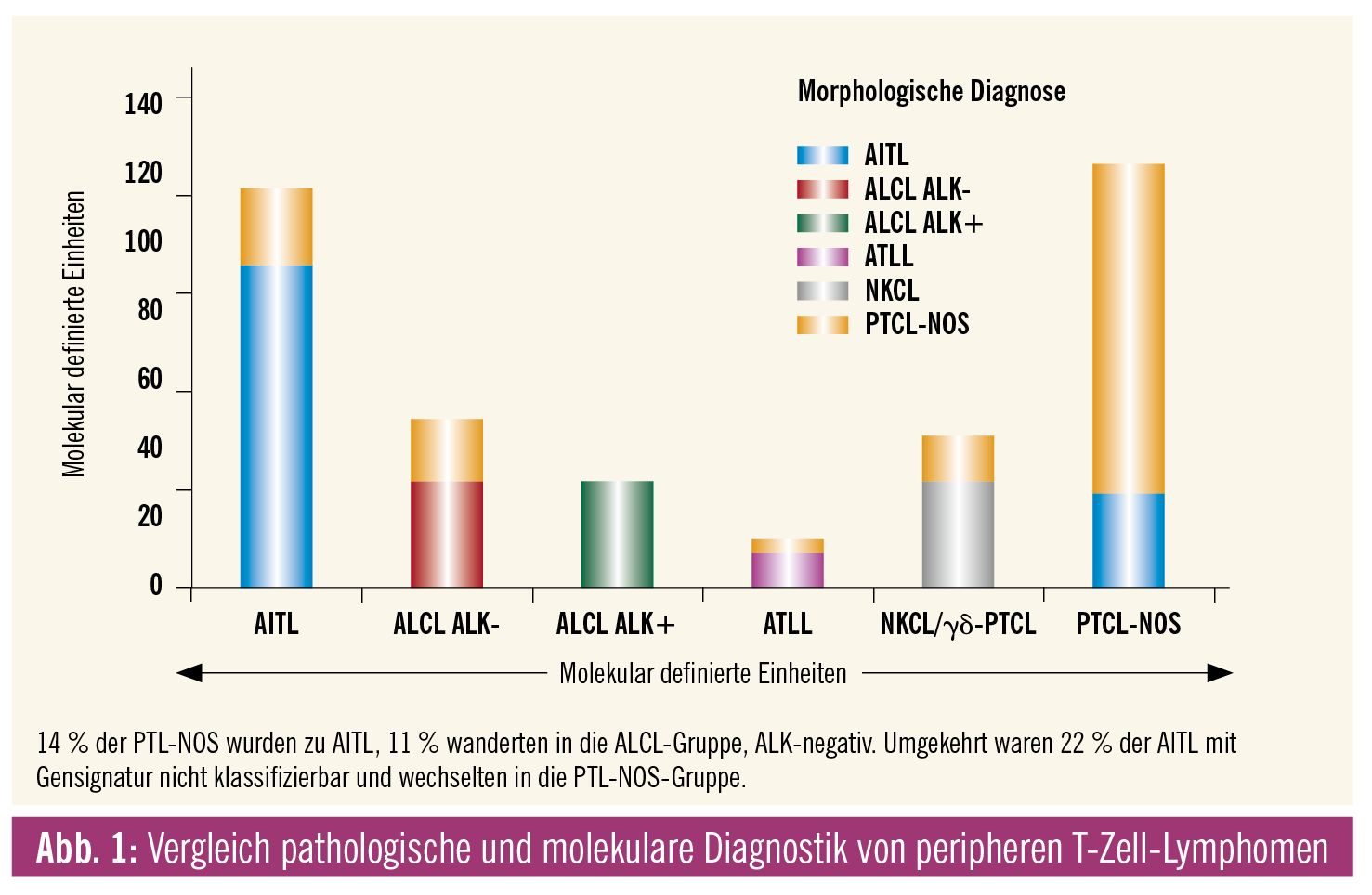
Bezüglich der Differenzialdiagnose zu anderen peripheren T-Zell-Lymphomen hat sich die Situation in den letzten Jahren etwas gebessert, da Genexpressions- und Sequenzierstudien den oben angeführten „Sammeltopf“ der PTL-NOS zumindest um einige falsch klassifizierte angioimmunoblastische T-Zell-Lymphome und um die neu eingeführte Kategorie der nodalen peripheren T-Zell-Lymphome mit T-follikulärem Helfer-Phänotyp bereinigen konnten (Abb. 1).3 Wichtigste und häufigste differenzialdiagnostische Problemzone ist die Abgrenzung zum anaplastischen großzelligen Lymphom, ALK-negativ. Diesbezüglich sind die uniforme starke CD30-Expression, die häufige Reaktivität für epitheliales Membranantigen (EMA) und der zytotoxische Immunphänotyp des anaplastischen großzelligen Lymphoms, ALK-negativ, richtungsweisend.
Die genetische Landschaft des peripheren T-Zell-Lymphoms, NOS
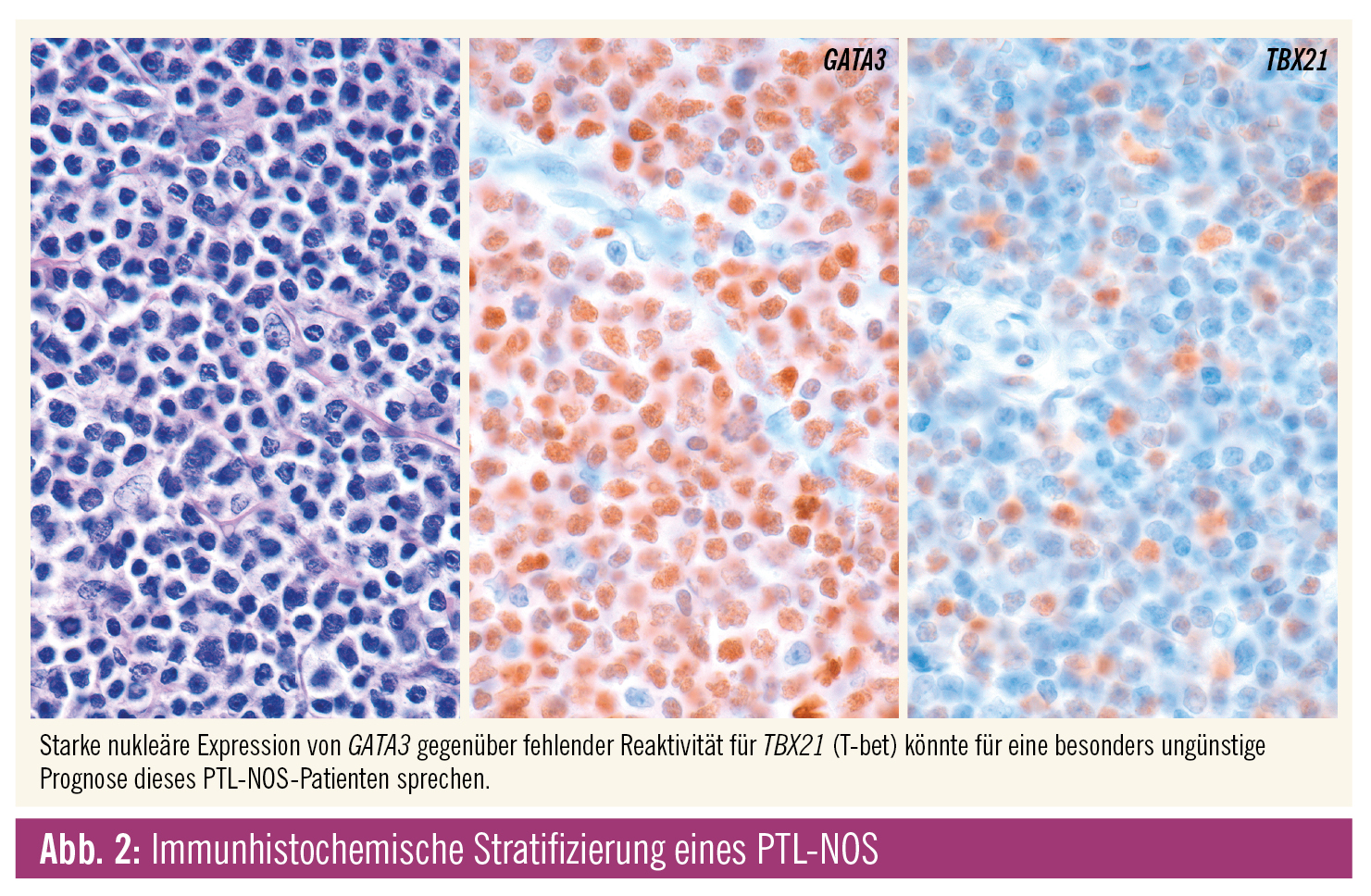
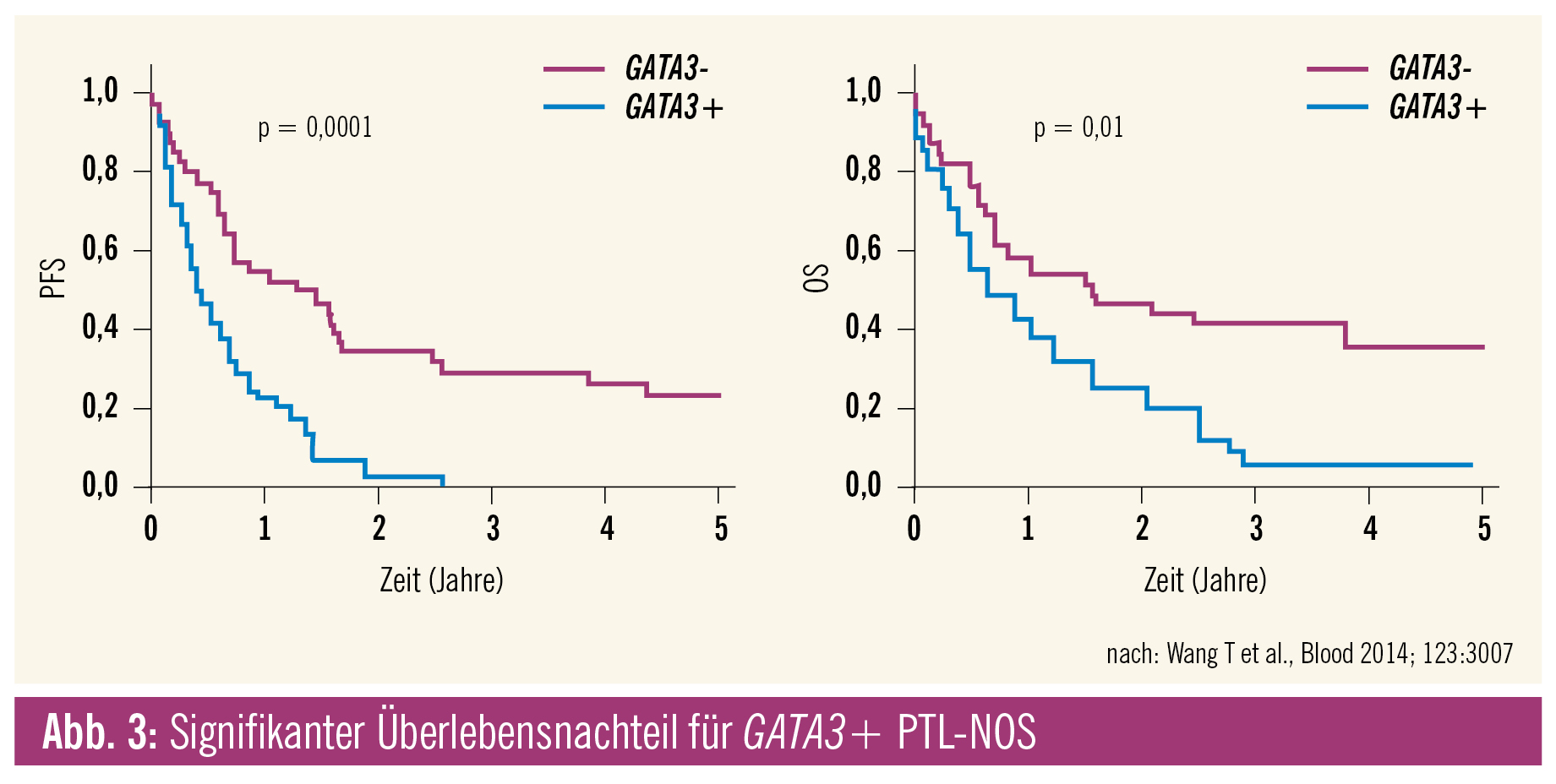
Genexpressionsstudien konnten PTL-NOS in zwei Hauptgruppen unterteilen, die signifikante Überlebensunterschiede aufweisen. Die eine Gruppe ist durch eine starke GATA3-Expression gekennzeichnet und mit einer hohen MYC und Proliferationssignatur assoziiert; die andere, quantitativ dominante (49 % aller PTL-NOS), durch eine TBX21 (T-bet)-Expression und den NF-B-Signalweg. Die 5-Jahres-Überlebensrate liegt anhand der die jeweilige Gruppe definierenden Gensignatur bei 38 % für die TBX21- und bei nur 19 % für die GATA3-Gruppe. Da die beiden Transkriptionsfaktoren GATA3 und TBX21 auch auf Proteinebene gut fassbar sind, könnte der Immunhistochemie prognostische Bedeutung zukommen. Das kleine PTL-NOS-Subset mit zytotoxischem Immunphänotyp dürfte Teil der TBX21-Gruppe sein.3
Da nur sehr wenige genetische Studien von PTL-NOS existieren, wurde eine zielgerichtete Sequenzierstudie von 237 Genen durchgeführt und es wurden häufige Mutationen (um 25 %) in epigenetischen Regulatoren (MLL28, KDM6A, MLL, TET1, TET2, DNMT3A) beobachtet.4 Etwa 10 % der PTL-NOS zeigen t(5;9)(q33;q22) mit dem T-Zell-Rezeptor-aktivierenden Fusionsprodukt ITK/SYK. In die T-Zell-Rezeptor-Signalübertragung involvierte VAV1-Fusionsprodukte wurden in 11 % der PTL-NOS gefunden, aber ebenso in 11 % der ALK-negativen großzelligen anaplastischen Lymphome.5 Entitätsüberlappende genetische Veränderungen kommen vor allem unter den drei peripheren T-Zell-Lymphomen angioimmunoblastisches T-Zell-Lymphom, PTL-NOS und ALK-negatives großzelliges anaplastisches Lymphom vor. Rekurrierende Mutationen von TET2, IDH2, DNMT3A, RHOA und CD28 wurden in PTL-NOS gefunden und halfen nach Korrelation mit Histologie und Immunphänotyp, die von follikulären Helfer-T-Zellen abstammenden Lymphome in der WHO-Klassifikation 2017 einer neuen Kategorie zuzuordnen („nodale periphere T-Zell-Lymphome mit T-follikulärem Helfer-Phänotyp“).6, 7
Die immunhistochemisch verifizierte CD30-Expression ist bei den PTL-NOS sehr unterschiedlich und korreliert sehr gut mit dem mRNA-Level. Genexpressionsstudien konnten zwei biologisch unterschiedliche Gruppen, nämlich PTL-NOS-CD30+ und PTL-NOS-CD30-, charakterisieren. Die Gruppe der PTL-NOS-CD30+ zeigt eine verminderte Expression von T-Zell-Differenzierungs-/Aktivierungsgenen (CD28, CD52, CD69, NFATc2) und T-Zell-Signaltransduktionsgenen (Tyrosinkinasen Lck, Fyn, Itk), hingegen eine verstärkte Expression von Transkriptionsfaktoren wie JunB und MUM1/IRF4. Dieses Expressionsmuster teilt die Gruppe der PTL-NOS-CD30+ mit den ALK-negativen großzelligen anaplastischen Lymphomen, während die Gruppe der PTL-NOS-CD30- ein inverses Expressionsprofil aufweist. Darüber hinaus nehmen die PTL-NOS-CD30- nicht nur auf molekularer Ebene eine Sonderstellung ein, sondern sie weisen mit einer medianen Gesamtüberlebenszeit von 10,5 Monaten auch die mit Abstand schlechteste Prognose auf.8
Trotz diagnostischem „fine-tuning“ der PTL-NOS gab es bislang leider keinen therapeutischen Durchbruch.
Resümee
Genexpressionssignaturen haben geholfen, den Sammeltopf der Kategorie PTL-NOS zu verkleinern. Die Grenze zum AITL und zu anderen von follikulären Helfer-T-Zellen abstammenden Lymphomen verläuft nun wesentlich schärfer. Die bekannte Grauzone zwischen PTL-NOS und anaplastischen Lymphomen, ALK-negativ, findet sich auch auf genetischer Ebene wieder, könnte aber auch gemeinsame therapeutische Targets enthalten. Die Aufteilung in die zwei Hauptgruppen GATA3+ versus TBX21+ identifiziert mit dem GATA3+-Kollektiv eine Hochrisikogruppe innerhalb der PTL-NOS.
1 Swerdlow SH et al. (eds.), IARC Lyon 2017; p345–p421
2 De Leval L et al., Haematologica 2015; 100:e361–e364
3 Iqbal J et al., Blood 2014; 123:2915–23
4 Schatz JH et al., Leukemia 2015; 29:237–41
5 Boddicker RL et al., Blood 2016; 128:1234–45
6 Sandell RF et al., Curr Oncol Rep 2017; 19:28
7 Lone W et al., Curr Hematol Malig Rep 2018; [ePub 27. Juni 2018]
8 Bisig B et al., Haematologica 2013; 98:1250–58
_Felicitas_Matern_opt.jpg)
AutorIn: Prim. Univ.-Prof. Dr. Andreas Chott
Institut für Pathologie und Mikrobiologie, Wilhelminenspital, Wien

Ursprünglich erschienen:
Patho 01|2018
Patho 01|2018
