





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Neue Targets in der Pharmakotherapie affektiver Erkrankungen
28. September 2012

Paradigmenwechsel in der Ätiologie affektiver Erkrankungen
Derzeitige psychopharmakologische Interventionen bei unipolarer Depression zielen auf die Beeinflussung monoaminerger Strukturen ab. Die sogenannte monoaminerge Hypothese der Depression besteht in der Annahme einer reduzierten Verfügbarkeit von Monoaminen, insbesondere von Serotonin und Noradrenalin im synaptischen Spalt. Folgerichtig zielen Antidepressiva hauptsächlich auf die Erhöhung dieser Amine an der Synapse ab, um so die Transmission zu beschleunigen.
Trotz 50-jähriger Forschung in diesem Bereich ist es allerdings bisher nicht gelungen, durch eine erste pharmakologische Intervention die Ansprechraten auf über 50 % zu heben. Nur 30 % der Patienten remittieren nach einem ersten Antidepressivum. Augenfällig ist auch die Latenzzeit zum Einsetzen der antidepressiven Wirksamkeit.
Die affektive Forschung der letzten 10 Jahre schwenkte den Focus in einem Ausmaß auf Glutamat und die damit verbundenen neuroplastischen Veränderungen, dass man von einem paradigmatischen Wechsel hin zu einer glutamatergen Hypothese affektiver Erkrankungen sprechen kann.
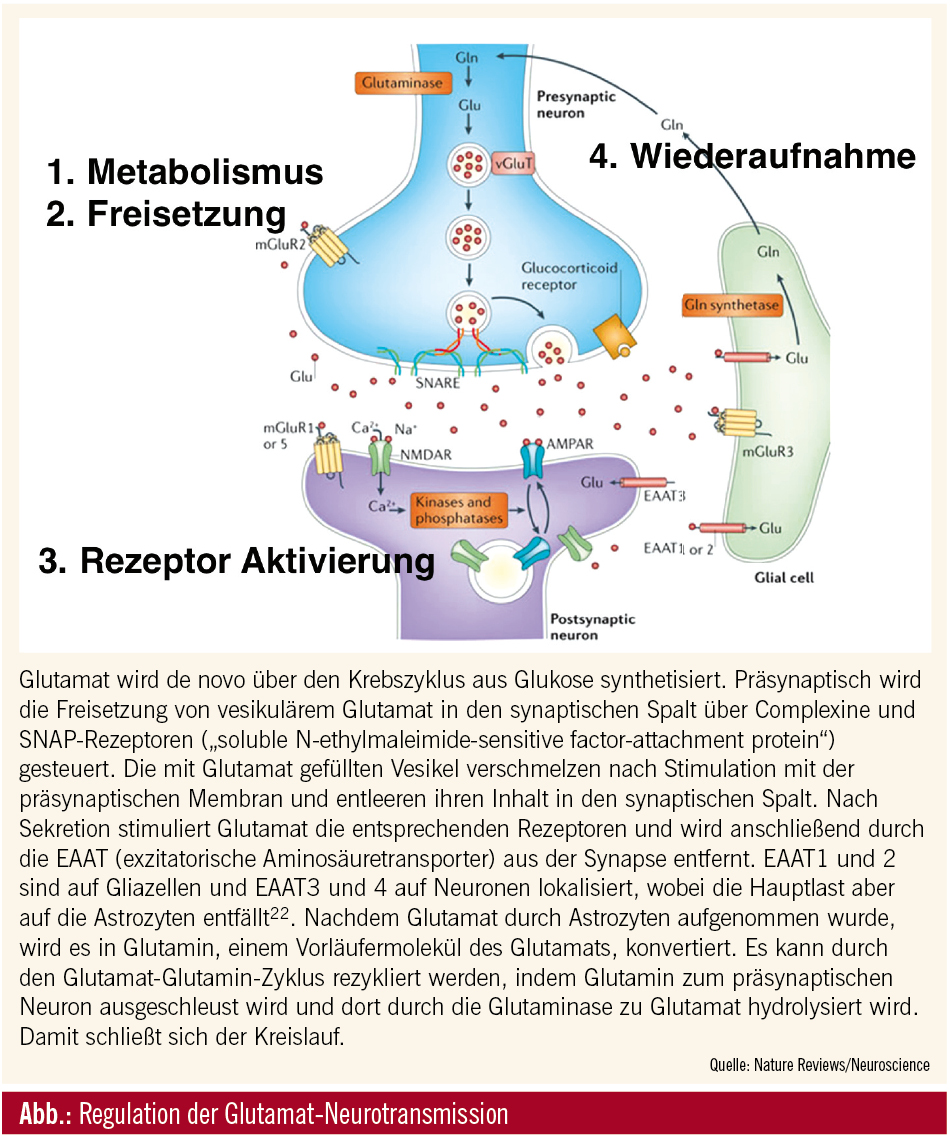
Das ZNS als glutamatgesteuerte Einheit
Die Aminosäure L-Glutamat ist der wichtigste exzitatorische Neurotransmitter im zentralen Nervensystem der Säugetiere. Der menschliche Neokortex stellt circa 85 % der gesamten Hirnmasse dar, wobei dessen Neuronen zu 80 % exzitatorisch glutamaterger Natur sind und 85 % aller Synapsen stellen. Glutamat ist im Gehirn an nahezu allen physiologischen Abläufen wie Kognition, Gedächtnis und Lernen beteiligt. Wichtige Aufgaben erfüllt es aber auch bei der Entwicklung des zentralen Nervensystems. So beeinflusst Glutamat zum Beispiel das Aussprossen neuronaler Fortsätze sowie die Migration von Neuronen.1
Glia und die neuronale Transmission
Die Anzahl der Gliazellen ist bei Säugetieren circa 10-mal höher als die der Neuronen, so dass die Rolle der Glia im Zusammenspiel aller Zellen, des ZNS wahrscheinlich eine größere ist, als bisher angenommen.2 Vor circa 10 Jahren wurde die Rolle der Astrozyten in der Aufrechterhaltung der glutamatergen Homöostase bekannt. Diese Aufgabe erfüllen sie durch Aufnahme und Abgabe von Glutamat.3
In dieser Zeit vollzog sich auch ein Wechsel in der Konzeption neuronaler Transmission. Synapsen wurden bis dahin als zweiteilig angesehen. Basierend auf der Beobachtung, dass Astrozyten, von allen Gliazellen, die bedeutendste Rolle in der Kommunikation zukommt, wurde der Begriff der dreiteiligen Synapse konzipiert.4 Sie besteht aus präsynaptischem Ende, postsynaptischer Membran sowie Astrozyten, die die synaptische Effizienz durch plastische Veränderungen verbessern.2 Die Aufrechterhaltung der Homöostase erfolgt durch die Entsorgung exzessiven extrazellulären Glutamats in die Astrozyten durch die exzitatorischen Glutamattransporter (EAAT) und die damit verbundene Vermeidung der Desensibilisierung der Rezeptoren. So wird die toxische Wirkung gesteigerter Glutamatmengen an den Rezeptoren (Exzitotoxizität) verhindert.
EAAT, VGLUT und xCT
Es wurden 3 Arten von Glutamattransportern identifiziert. Neben den EAAT, deren Funktion auf spannungsabhängigen Natriumkanälen beruht, konnten noch die sogenannten VGLUT (vesikuläre Glutamattransporter) sowie die xCT (Cystin-Glutamat-Antiporter) identifiziert werden. Diese beruhen im Gegensatz zu den EAAT auf anderen Mechanismen.
Nach Sekretion stimuliert Glutamat die entsprechenden Rezeptoren und wird anschließend durch die EAAT (exzitatorische Aminosäuretransporter) aus der Synapse entfernt.
Die VGLUT sind auf den Membranen glutamathaltiger Vesikeln situiert. Es existieren 4 Typen. VGLUT 1–3 respektiv SLC17A6–8 und der neue Glutamat-Aspartat-Transporter Sialin. Diese Transporter packen Glutamat in synaptische Vesikel.
Die Cystin-Glutamat-Antiporter (xCT) sind auf Plasmamembranen lokalisiert und bestehen aus 2 Proteinkomponenten. Das xCT-Protein ist für den intrazellulären Transport eines Moleküls Cystin, der oxygenierten Form des Cysteins, im Austausch mit einem Molekul Glutamat in den extrazellulären Raum verantwortlich. Bisher war man der Meinung, dass das xC-System ein reines Versorgungssystem der Zellen mit Cystein zur Synthese des Antioxydans Glutathion sei. Neueste Evidenz deutet darauf hin, dass das xC-System ein von der Glutathionsynthese unabhängiges Redoxsystem darstellt.5
Glutamat wirkt über verschiedene Rezeptor-Typen
Glutamat wirkt über 2 Typen von Rezeptoren, welche auf fast allen Neuronen lokalisiert sind. Einige dieser Rezeptoren werden auch von Gliazellen exprimiert.
Die 2 Arten von Glutamatrezeptoren sind einerseits ionotrope Glutamatrezeptoren (iGluR), die eine schnelle synaptische Übertragung mediieren, und andererseits die metabotropen mGluR.
iGlu-Rezeptoren
Die iGlu-Rezeptoren, auch Ionenkanäle genannt, sind porenbildende Transmembranproteine, die elektrisch geladenen Teilchen, also Ionen, das Durchqueren von Biomembranen ermöglichen. Der Transport erfolgt dabei entlang des bestehenden elektrochemischen Gradienten (dem Konzentrations- und Potenzialgefälle). iGlu-Rezeptoren sind NMDA-, AMPA- und KA-Rezeptoren.
Die NMDA-Rezeptoren (N-Methyl-D-Aspartat-Rezeptor; NMDAR) setzen sich als Tetramer aus den Untereinheiten NR1, NR2 (NR2A–NR2D) und NR3 (NR3A und NR3B) zusammen. Für eine Aktivierung des NMDA-Rezeptors ist nicht nur die Bindung von Glutamat, sondern auch die des Koagonisten Glyzin erforderlich. Der Bindungsort für NMDA-Liganden scheint an NR2 lokalisiert zu sein, wobei die Subeinheit NK2 das Glyzin binden soll. Die Stimulation der NMDAR induziert einen Ca++-Einstrom mit nachfolgender Genexpression über Aktivierung der sogenannten Signal-transmissionskopplung.
Die AMPA-Rezeptoren (α-Amino-3-hydroxy-5-methyl-4-isoxazol-Propionsäure-Rezeptoren; AMPAR) bestehen aus den Untereinheiten iGluR1 bis iGluR4. An reifen Synapsen werden AMPA-Rezeptoren oft neben NMDA-Rezeptoren koexprimiert, wobei im Zusammenspiel diese auch bei Aufgaben der neuronalen Plastizität wie Lernen und Gedächtnis sowie bei der Neuroprotektion eine Rolle übernehmen. Die AMPA-Rezeptoren sind für die schnelle synaptische Erregung durch Glutamat verantwortlich. Bei Aktivierung öffnen AMPAR die Poren und lassen so Natrium in die Zelle fließen, was dementsprechend die Depolarisation der Zellmembran zur Folge hat.
Der NMDAR-bedingte Ca++-Influx in das postsynaptische Neuron initiiert eine Signalkaskade. Diese Kaskade hat eine schnelle Einfügung von iGluR1-AMPAR in die Synapse zur Folge, um so die postsynaptische Erregung zu erhöhen.
Die KA-Rezeptoren (Kainat-Rezeptoren) bestehen aus den Untereinheiten GluR5 bis 7, KA-1 und KA-2.
Metabotrope Rezeptoren (mGlu)
Die zweite Art glutamaterger Rezeptorstimulation sind die metabotropen Rezeptoren (mGlu), welche an G-Proteine gekoppelt sind. Diese haben eine modulierende Wirkung auf das ZNS, d. h. dass sie mehr Modulation als direkte Stimulation des glutamatergen Systems ausüben. Sie werden in drei Gruppen unterteilt: Gruppe I (mGluR1, mGluR5), Gruppe II (mGluR2 und mGluR3) und Gruppe III (mGluR4, mGluR6 bis mGluR8).
Momentan scheinen die potentesten antidepressiven Interventionen die negativen allosterischen Modulatoren der Gruppe I (mGlu1 und mGlu5) sowie die positiven allosterischen Modulatoren der Gruppe II (mGlu2 und mGlu3) sowie der Gruppe III (mGlu4/7/8) zu sein. Die Bezeichnung G-Protein steht für Guaninnukleotid-bindendes Protein oder GTP-bindendes Protein. G-Proteine besetzen eine Schlüsselposition in der Signalweiterleitung (Signaltransduktion) zwischen Rezeptor und Second-Messenger-Systemen. Beispiele hierfür sind Adenylylcyclasen, G-Protein-gekoppelte Rezeptorkinasen (GRK) und Phospholipasen.
Glutamat und die Pathogenese affektiver Erkrankungen
Aufgrund der multiplen intrazellulären Funktionen ist die intrazelluläre Glutamatkonzentration der Neuronen des ZNS sehr hoch. Diese hohen Konzentrationen erfordern genau abgestimmte metabolische Prozesse, da zu hohes extrazelluläres Glutamat über die sogenannte glutamaterge Exzitotoxizität, d. h. einer chronischen Dauerstimulation der Rezeptoren, zu einer Schädigung der Neuronen führen würde. Die regulativen Prozesse führen im Normalfall zu einer Reduktion extrazellulären Glutamats, um so eine optimale Transmission zu gewährleisten.
Es hat sich in den letzten Jahren die Erkenntnis über eine differenzielle Wirkung der Aktivierung der glutamatergen NMDA-Rezeptoren herausgestellt. Intra- und extrasynaptische NMDA-Rezeptoren haben unterschiedliche Funktionen. War man früher der Ansicht, dass die toxische Wirkung einer chronischen Rezeptoraktivierung durch die Menge des einströmenden Ca++ bedingt ist, weiß man heute, dass der CA++-Einstrom, welcher durch eine intensive synaptische NMDA-Aktivierung bedingt ist, der Apoptose entgegenwirkt.
Nur bei Erregung synaptischer NMDA-Rezeptoren kommt es zu einer dauerhaften Phosphorylierung und damit Aktivierung des Transkriptionsfaktors CREB („cAMP response element-binding protein“), der wiederum mit einer gesteigerten Synthese des „brain-derived neurotrophic factor“ (BDNF) assoziiert ist. BDNF ist ein zentrales Element der neuronalen Plastizität und übt trophische Effekte auf Neurone aus. Experimentelle und klinische Daten zeigen bei Depression eine reduzierte Expression von BDNF, die sich durch Behandlung mit Antidepressiva signifikant erhöhte. Extrasynaptische NMDA-Rezeptoren aktivieren CREB nur unzureichend, was durch eine insuffiziente Expression von BDNF und antiapoptotischer Gene eine vermehrte Apoptosefähigkeit neuronaler Zellen bedingt und so mitochondriale Dysfunktionen und Zelltod zur Folge haben.6
Akuter Stress
Akuter Stress und Glukokortikoide erhöhen extrazelluläre Glutamatlevel, z. B. im Hippokampus, in der Amygdala oder im präfrontalen Kortex (PFC).
Akuter Stress induziert eine akute Glukokortikoid-Rezeptor-Aktivierung (GR-Aktivierung) sowie eine Akkumulation von SNARE-Komplexen in der präsynaptischen Membran, die eine rapide Steigerung depolarisationsbedingter Ausschüttung von Glutamat zur Folge haben. Diese Effekte konnten durch die Gabe von Antidepressiva verhindert werden. Akuter Stress erhöht auch die glutamaterge synaptische Transmission durch Steigerung der Aktivität der NMDAR und AMPAR durch gesteigerte Rezeptorenaussprossung in Neuronen des präfrontalen Kortex.
Negative Effekte von akutem Stress auf Gliazellen, und zwar auf den Glutamattransporter EAAT2, sind beschrieben worden. Bei reduzierter Aktivität der EAAT wird vermindert Glutamat aus den Synapsen nach intrazellulär evakuiert. Dies ist besonders relevant, wenn durch Stress induzierte erhöhte extrazelluläre Glutamatwerte vorliegen. Post-Mortem-Untersuchungen bei Depressiven konnten eine Reduktion der glialen Zelldichte in Regionen, die für die emotionale Verarbeitung verantwortlich sind, z. B. Amygdala und dem subgenualen präfrontalen Kortex, berichten. Ein regionaler Verlust von Gliazellen zeichnet für eine reduzierte Aufnahme von Glutamat verantwortlich. Diese hat wiederum eine Erhöhung von extrazellulärem Glutamat zur Folge.7
Diese Befunde passen gut zum postulierten Konzept der Depression als hyperglutamaterger Zustand, wonach Gliazellen als Teil der dreiteiligen Synapsen eine maßgebliche Rolle als Aufnahmepuffer für freies Glutamat spielen.
Chronischer Stress
Chronischer Stress inhibiert die synaptische Plastizität in dem Sinn, dass die „long-term potentiation“ (LTP) in den Thalamus-PFC- und Hippocampus-PFC-Loops heruntergefahren wird. Es wird vermutet, dass chronischer Stress – im Vergleich zu akutem Stress – eine dauerhaft erhöhte Glutamatausschüttung zur Folge hat.
Oxidative Glutamattoxizität
Eine weitere Erklärung für die nervenzellschädigende Wirkung des Glutamats gibt die Hypothese der sogenannten oxidativen Glutamattoxizität. Hohe extrazelluläre Glutamatkonzentrationen inhibieren den Cystein-Glutamat-Antiporter (xCT), so dass die Aufnahme von Cystein und konsekutiv die Glutathionsynthese stark vermindert wird. In Folge des oxidativen Stresses kommt es zum Zelluntergang.
Neue Substanzen in der Therapie affektiver Erkrankungen
Neue neurobiologische Interventionen zur Entstehung schwerer rezidivierender affektiver Erkrankungen beinhalten sowohl Veränderungen der intrazellulären Signal-Transmissions-Kopplung als auch Beeinflussungen der zellulären Plastizität.7 Die Hypothese gestörter Neuroplastizität bei affektiven Erkrankungen postuliert eine durch Stress erhöhte abnorme Erhöhung exzitatorischer Aminosäuren in bestimmten Hirnarealen und dadurch bedingte morphologische/funktionelle Veränderungen.
Circa 20 präklinische Studien wurden bisher zur antidepressiven Wirksamkeit antiglutamaterger Substanzen publiziert. Die meisten dieser präklinischen Studien bedienten sich des sogenannten Forced-Swim-Tests (FST) als Tiermodell für depressives Verhalten. Aber auch eine Abwandlung vom FST, der Tail-Suspension-Test (TST), wurde als Verhaltensparadigma verwendet. In diesen Tests wird das passive Coping als Korrelat für depressives Verhalten betrachtet. Diese Immobilität vermindert sich nach Gabe von antidepressiv wirksamen Substanzen.8
NMDA-Rezeptor-Antagonisten
Es sind besonders NMDA-Antagonisten, wie MK-801, AP-7 und Neramexan, die in den beiden Tests, dem FST und dem TST, signifikante Ergebnisse brachten.
Ketamin gehört zu den bekanntesten nichtkompetitiven NMDA-Rezeptor-Antagonisten mit hoher Rezeptoraffinität. Viele Studien mit positiven Effekten liegen auch in beiden vorhin erwähnten präklinischen Verhaltensparadigmen vor. Ketamin konnte robuste und schnell einsetzende antidepressive Effekte zeigen. Diese Effekte dürften aber zumindest teilweise durch die der NMDAR-Antagonisten mediierten Effekte auf die Erhöhung der Exzitation postsynaptischer AMPA-Rezeptoren durch gesteigerte Expression an der Membran zustandekommen.9
Die beiden am meisten zitierten klinischen Studien zu den antidepressiven Wirkungen von Ketamin sind die von Berman et al.10 und Zarate et al.11 Es sind dies randomisierte, placebokontrollierte, doppelblinde Cross-over-Studien mit therapieresistenten depressiven Patienten. Bereits nach einer einzelnen Ketamingabe von 0,5 mg/kg KG und innerhalb von Stunden war in diesen Untersuchungen ein im Vergleich zu Placebo signifikanter antidepressiver Effekt (Response) zu beobachten. Die Response – teilweise sogar Remission – hielt bis zu einer Woche lang, in einigen Fällen länger. Bis auf eine vorübergehende kognitive Störung und Wahrnehmungsveränderungen zeigte Ketamin eine insgesamt gute Verträglichkeit.
In der Studie von Zarate et al.11 konnte eine signifikante Response bereits nach 2 Stunden nach i. v. Ketaminadministration im Vergleich zu Placebo dokumentiert werden. Nach einem Tag zeigten 29 % der Patienten eine Remission und 71 % eine Response. Bei insgesamt 35 % blieb der Effekt einer Response nach Einzelgabe für mindestens eine Woche erhalten. Nebenwirkungen waren bis zu 110 min nach Ketamininfusion in Form von Wahrnehmungsstörungen, Verwirrtheit, kognitiver Störung, Blutdruckerhöhung, Euphorie, Schwindel und Libidosteigerung feststellbar.
Orales S-Ketamin wurde als Add-on zu einer Standardbehandlung mit Antidepressiva bei 4 depressiven Patienten eingesetzt. S-Ketamin zeigt geringere psychotomimetische Nebenwirkungen, aber auch Wirkungen als sein Enantiomer R-Ketamin bzw. als das Racemat aus S-/R-Ketamin. Orales S-Ketamin wurde ohne Nebenwirkungen gut vertragen. Obschon seit 1990 einige Studien publiziert wurden, fehlen allerdings immer noch große randomisierte, kontrollierte Studien. Nicht zuletzt aufgrund der potenziellen toxischen Nebenwirkungen von Ketamin und bestehender Unklarheit bezüglich phasenprophylaktischer Wirksamkeit ist derzeit an einen Routineeinsatz dieses Substanz nicht zu denken.
Memantin ist ein mit geringer Rezeptoraffinität versehener nichtkompetitiver NMDA-Rezeptor-Antagonist. Die Substanz wirkte sich in präklinischen Studien auf die Reduktion der Immobilitätszeit günstig aus. Memantin hat Eingang in die Therapie bei mittel- und schwergradigen Formen der Alzheimer-Demenz gefunden.
Memantin war in einer Studie antidepressiv wirksam, ein Effekt, der in einer früheren doppelblinden, randomisierten und placebokontrollierten Depressionsstudie nicht gezeigt werden konnte.12 In einer Studie mit Alkoholabhängigen mit komorbider Depression zeigte Memantin eine dem Escitalopram vergleichbare Wirksamkeit. Die Studie war allerdings ohne Placebovergleich.
Ifenprodil, Traxoprodil: Die neuesten Entwicklungen im Bereich der antiglutamatergen Substanzen beinhalten bereits Antagonisten von NMDA-Rezeptor-Untereinheiten (z. B. selektive NR2B-Antagonisten: Ifenprodil, Traxoprodil), die in einer doppelblinden, randomisierten placebokontrollierten Studie antidepressive Wirksamkeit gezeigt haben. Medikamente, die die Glutamatfreisetzung modulieren, wie beispielsweise Lamotrigin, zeigten antidepressive Wirksamkeit.
Riluzol ist eine Substanz, die v. a. über die Hemmung spannungsabhängiger Natriumkanäle die Freisetzung von Glutamat antagonisiert bzw. den Einstrom extrazellulären Glutamats in die Zelle verstärkt. Riluzol ist bisher nur in der Therapie der amyotrophen Lateralsklerose zugelassen.
Das Modell des „chronic unpredictable stress“ verursacht bei den Tieren Verhaltensänderungen wie z. B. eine reduzierte Präferenz für Zuckerlösung, das als Zeichen für Anhedonie interpretiert wird. Diese Reduktion der Präferenz konnte durch die chronische Gabe des antiglutamaterg wirksamen Riluzol rückgängig gemacht werden.13 Die Substanz zeigte antidepressive Effekte in der Monotherapie14 und als Augmentationstherapie bei therapieresistenter unipolarer Depression.15 Es zeigte in klinischen Studien auch antidepressive Wirksamkeit als Monotherapie und auch als Augmentation zu Lithium bei bipolarer Depression. Allerdings wurde in einer kürzlich erschienenen placebokontrollierten Studie unter Riluzol eine höhere Rückfallrate in die Depression beobachtet als unter Placebo.
Ceftriaxon, ein β-Lactam-Antibiotikum aus der Gruppe der Cephalosporine der 3. Generation, erleichtert den Transport von Glutamat in Gliazellen. Es scheint selektiv das Gen für den EAAT2 in Gliazellen hochzuregulieren, so dass die Ausschleusung von Glutamat aus glutamatergen Synapsen begünstigt wird.16 Es hat sowohl neuroprotektive wie auch antidepressive Effekte.13
Modulatoren der mGlu-Rezeptoren (mGluR)
Substanzen, die auf metabotrope Glutamatrezeptoren (mGluR) einwirken, sind jene mit vielversprechenden Daten in der Therapie psychiatrischer Erkrankungen. Auch wenn sich die Forschung in einem sehr frühen Stadium bewegt und es derzeit noch kein einziges klinisch zugelassenes Präparat gibt, korrelieren die präklinischen Daten gut mit dem Einsatz beim Menschen.
Gruppe I: Antagonisten der metabotropen mGluR5-Rezeptoren (Gruppe I) konnten antidepressive und anxiolytische Effekte in präklinischen Studien zeigen. Es konnte gezeigt werden, dass Antagonisten des mGluR1 und des mGluR5 sowohl im FST als auch im TST die Immobilitätszeit der Tiere reduzierten. Analog zu diesen Antagonisten-Studien weisen Knock-out-Mäuse für mGluR5 eine reduzierte Immobilitätszeit im FST auf.
mGluR5-Rezeptoren-Antagonisten können auch über Wirkung auf die auf Gliazellen lokalisierten 1-mGlu-Rezeptoren antidepressive Effekte induzieren, indem sie die hypothesierte glutamaterge Überstimulation extrasynaptischer NMDA-Rezeptoren reduzieren.7 Konsistent mit dieser Theorie ist die erhöhte mRNA-BDNF-Expression im Ratten-Hippokampus nach chronischer Applikation des mGluR5-Antagonisten MPEP.
Aus der Gruppe II der metabotropen Glutamat-Rezeptor-Antagonisten und negativen allosterischen Modulatoren wie beispielsweise MGS0039 und LY341495 erwiesen sich sowohl mGluR2- als auch mGluR3-Antagonisten im FST und TST antidepressiv wirksam. Zugleich konnte damit der Proof-of-Concept-Beweis erbracht werden, dass eine Dämpfung dieser Rezeptorengruppe und der dadurch bedingten Reduktion präsynaptischer Glutamatfreisetzung einen antidepressiven Effekt induziert.17
Aus der Gruppe III der metabotropen Glutamat-Rezeptoren haben sich im FST Agonisten an den Rezeptoren mGluR4, mGluR7 und mGluR8 als antidepressiv wirksam erwiesen. Neueste präklinische Daten suggerieren, dass die Latenz des Eintretens der antidepressiven Wirkung bei der Zugabe eines mGlu2/3-Liganden zu einem bestenden Antidepressivum deutlich verkürzt wird. Derzeit sind einige Substanzen in klinischer Phase-II-Erprobung.18
AMPAR-Modulatoren (AMPA-kine)
AMPA-Rezeptor-Modulatoren sind keine Agonisten im klassischen Sinne, sondern sie verlangsamen die Rate von Rezeptordesensitivierung und/oder -deaktivierung im Beisein eines Agonisten. Chourbaji et al.19 stellten Knock-out-Mäuse für die Haupteinheit des AMPA-Rezeptors (GluR-A) als transgenes Tiermodell für die Depression vor; diese Mäuse zeigen eine erhöhte erlernte Hilflosigkeit, reduzierte zentralnervöse Serotonin- und Noradrenalinkonzentrationen, erhöhte Glutamatkonzentrationen und eine erhöhte Expression von NMDA-Rezeptoren.
Die Ergebnisse machen deutlich, dass Veränderungen an AMPA-Rezeptoren in der Pathophysiologie der Depression involviert sein und gleichzeitig therapeutisch genutzt werden könnten. Substanzen mit Wirkung auf AMPAR, speziell solche, die die Aktivierung fördern, wie beispielsweise LY392098, zeigen nootrope und antidepressive Effekte bei Nagern. Die Vorbehandlung von Mäusen mit dem AMPA-Rezeptor-Antagonisten NBQX schwächte die ketaminassoziierten antidepressiven Effekte in Tiermodellen ab, was eine besondere Bedeutung von AMPA-Rezeptoren bei den ketaminassoziierten antidepressiven Effekten in den Vordergrund rücken lässt.
Andere glutamaterge Substanzen
Amantadin: Auch andere antiglutamaterge Substanzen wie Amantadin konnten antidepressive Wirksamkeit im FST zeigen.
N-Azetylcystein: Ein neuer antidepressiver Funktionsmechanimus fokussiert auf den Glutamat-Cystein-Antiporter (xCT). N-Azetylcystein passiert die Blut-Hirn-Schranke und erhöht die Gliazellexpression des Glutamat-Cystein-Antiporters, einem Cystein-Transport-Protein, welches Glutamat gegen Cystein im Modus 1 : 1 aus der Synapse entfernt. N-Azetylcystein erwies sich in einer doppelblinden, randomisierten, placebokontrollierten Studie sicher und effektiv als Augmentation bei der Depression im Rahmen einer bipolaren affektiven Störung.20
Andere neue Strategien
In der Depression sind Endocannabinoide im präfrontalen Kortex und Hippokampus von Nagern reduziert. Die Stimulation der Cannabinoid-1-Rezeptoren (CB1) zeigte anxiolytische und antidepressivähnliche Effekte im Tiermodell, so dass diese Strategie kürzlich auch als zukunftsweisend für die klinische Verwendung eingestuft wurde.21
Zu Bitopertin, einem Glycin-Wiederaufnahme-Hemmer, der in der Therapie der Schizophrenie als Add-on-Strategie bereits Wirksamkeit zeigte, werden derzeit Phase-II-Studien, sowohl zur Behandlung der akuten Depression und Manie als auch zur Phasenprophylaxe in der bipolaren Störung, erarbeitet.
Resümee
Aufgrund einiger inkonziser Aspekte der monoaminergen Theorie der Entstehung affektiver Erkrankungen kam es in den letzten Jahren zu einem Paradigmenwechsel hin zum glutamatergen System und der damit verbundenen Fähigkeit plastischer Veränderungen des ZNS. Die Hypothese gestörter Neuroplastizität bei affektiven Erkrankungen postuliert eine durch Stress erhöhte abnorme Erhöhung exzitatorischer Aminosäuren in bestimmten Hirnarealen und dadurch bedingte morphologische und funktionelle Veränderungen.
Zu hohes extrazelluläres Glutamat führt über die sogenannte glutamaterge Exzitotoxizität, d. h. einer chronischen Dauerstimulation der Rezeptoren, zu einer Schädigung der Neuronen. Die regulativen Prozesse führen im Normalfall zu einer Reduktion extrazellulären Glutamats, um so eine optimale Transmission zu gewährleisten. Die Aufrechterhaltung dieser Homöostase erfolgt durch die Entsorgung exzessiven extrazellulären Glutamats in die Astrozyten durch die Plasmamembran-Glutamat-Transporter (EAAT). Auch eine reduzierte Glutamatfreisetzung aus den Vesikeln durch SNARE-Proteine sowie ein intakter Austausch von Glutamat mit Cystin durch die xCT (Cystin-Glutamat-Antiporter) können diese Homöostase aufrechterhalten.
Die Blockade von NMDA-Rezeptoren, wie durch Ketamin, ist in präklinischen, aber auch in klinischen Studien durch eine robuste und schnell wirksame antidepressive Wirksamkeit gekennzeichnet. Es fehlen aber immer noch große kontrollierte Studien, die die Frage der Langzeitanwendung sowie die der Nebenwirkungen zuverlässig beantworten können. Obschon es derzeit kein einziges zugelassenes Medikament auf dem Markt gibt, scheinen die vielversprechendsten antidepressiven Interventionen die negativen allosterischen Modulatoren der glutamatergen Rezeptorengruppe I (mGlu1 und mGlu5) sowie die positiven allosterischen Modulatoren der Gruppe II (mGlu2 und mGlu3) und der Gruppe III (mGlu4/7/8) zu sein.
Phase-II-Studien zielen derzeit auch auf eine Add-on-Gabe zu bestehender antidepressiver Medikation hin, um so bei wenig Nebenwirkung die antidepressive Wirksamkeit zu beschleunigen. Aber auch die sogenannten AMPA-kine sowie nichtglutamaterge Strategien, wie die Modulation des Endocannabinoidsystems, zählen zu den neuen pharmakologischen Strategien bei affektiven Erkrankungen.
1 Komuro H, Rakic P, Modulation of neuronal migration by NMDA receptors. Science 1993; 260:95–97.
2 Volterra A, Meldolesi J, Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci 2005; 6:626–640.
3 Schousboe A, Role of astrocytes in the maintenance and modulation of glutamatergic and GABAergic neurotransmission. Neurochem Res 2003; 28:347–352.
4 Smit AB, Syed NI, Schaap D, van Minnen J, Klumperman J, Kits KS, Lodder H, van der Schors RC, van Elk R, Sorgedrager B, Brejc K, Sixma TK, Geraerts WP, A glia-derived acetylcholine-binding protein that modulates synaptic transmission. Nature 2001; 411:261–268.
5 Conrad M, Sato H, The oxidative stress-inducible cystine/glutamate antiporter, system x (c) ( ): cystine supplier and beyond. Amino Acids 2012; 42(1):231–46.
6 Hardingham GE, Bading H, Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nature Rev Neurosci 2010; 11:682–696.
7 Sanacora G, Zarate CAJ, Krystal JH et al., Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nature Rev Drug Discov 2008; 7(5):426–437.
8 Pittenger C, Duman RS, Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology 2008; 33(1):88–109.
9 Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS, mTOR–dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010; 329:959–964.
10 Berman RM, Cappiello A, Anand A et al., Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 2000; 47:351–354.
11 Zarate CA Jr, Singh JB, Carlson PJ et al., A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 2006; 63:856–864.
12 Zarate CA Jr, Singh JB, Quiroz JA et al., A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am J Psychiatry 2006; 163:153–155.
13 Banasr M, Chowdhury GM, Terwilliger R et al., Glial pathology in an animal model of depression: reversal of stress-induced cellular, metabolic and behavioural deficits by the glutamate-modulating drug riluzole. Mol Psychiatry 2010; 15(5):501–11.
14 Zarate CA Jr, Payne JL, Quiroz J et al., An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry 2004; 161:171–174.
15 Sanacora G, Kendell SF, Fenton L et al., Riluzole augmentation for treatment-resistant depression. Am J Psychiatry 2004; 161:2132.
16 Rothstein JD, Patel S, Regan MR et al., Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 2005; 433(7021):73–7.
17 Koike H, Iijima M, Chaki S, Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav Brain Res 2011; 224:107–111.
18 Matrisciano F et al., Group-II metabotropic glutamate receptor ligands as adjunctive drugs in the treatment of depression: a new strategy to shorten the latency of antidepressant medication? Molecular Psychiatry 2007; 12:704–706.
19 Chourbaji S, Vogt MA, Fumagalli F et al., AMPA receptor subunit 1 [GluR-A] knockout mice model the glutamate hypothesis of depression. FASEB J 2008; 22:3129–3134.
20 Berk M, Copolov DL, Dean O et al., N-acetyl cysteine for depressive symptoms in bipolar disorder – a double-blind randomized placebocontrolled trial. Biol Psychiatry 2008; 64:468–475.
21 Valverde O, Torrens M, CB1 receptor-deficient mice as a model for depression. Neuroscience 2012; 204:193–206.
22 Pittenger C, Sanacora G, Krystal JH, The NMDA receptor as a therapeutic target in major depressive disorder. CNS Neurol Disord Drug Targets 2007; 6(2):101–15.

Ursprünglich erschienen:
SP 03|2012
SP 03|2012