





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Therapie des metastasierten hormonsensitiven Prostatakarzinoms (mHSPC)
Schon vor mittlerweile mehr als 5 Jahren haben die CHAARTED- und STAMPEDE-Studien gezeigt, dass eine alleinige Androgendeprivationstherapie (ADT) beim primär metastasierten hormonsensitiven Prostatakarzinom nicht ausreichend ist. Die Ergebnisse dieser beiden Studien führten zur Zulassungserweiterung von Docetaxel (6 Zyklen, 75 mg/KOF, 3-wöchentlich) und Abirateron (plus 5 mg Prednison) beim mHSPC.
Die finale Überlebensanalyse der LATITUDE-Studie zu Abirateron plus Prednison wurde 2019 veröffentlicht. Auch in der finalen Analyse konnte bestätigt werden1, dass die Kombination von Abirateron und Prednison mit einer ADT bei Männern mit neu diagnostiziertem metastasiertem kastrationssensitivem Prostatakrebs (mHSPC) mit hohem Risiko zu einem signifikant längeren Gesamtüberleben (OS) als Placebo plus ADT führt.
Definition Hochrisiko nach LATITUDE (zwei der folgenden drei Hochrisikokriterien):
- Gleason Score ≥ 8
- Vorhandensein von ≥ 3 Läsionen im Knochenszintigramm
- und/oder das Vorhandensein von messbaren viszeralen Läsionen
Drei weitere Studien wurden 2019 präsentiert, sie zeigen, dass Enzalutamid (ENZAMET-Studie, ARCHES-Studie) respektive Apalutamid (TITAN-Studie) ebenfalls zu einer Verlängerung der Progressionszeit und des Gesamtüberlebens beim mHSPC führen.
ENZAMET-Studie (Enzalutamid)
Die Rationale für ENZAMET (Enzalutamide in First Line Androgen Deprivation Therapy for Metastatic Prostate Cancer) waren die Daten hinsichtlich der frühzeitigen Ergänzung einer ADT mit Docetaxel oder Abirateron (s. o.), durch das Männer mit mHSPC klinisch relevant von einem verlängerten Gesamtüberleben profitierten.
Inwieweit dieses auch bei einer Ergänzung der ADT mit Enzalutamid +/– Docetaxel zutrifft, war bislang nicht untersucht worden. Diese Fragestellung sollte in ENZAMET2 durch Zusatz von Enzalutamid zur Erstlinienbehandlung aus Testosteronsuppression +/– frühzeitige Docetaxel-Chemotherapie geklärt werden.
Die Phase-III-Studie inkludierte 1.125 Männer in 83 Zentren auf zwei Gruppen verteilt mit Testosteronsuppression plus entweder Enzalutamid (Enza-Gruppe; n = 563) oder einem standardmäßigen nichtsteroidalen Antiandrogen (Standard- Gruppe; n = 562). Als primärer Endpunkt war das Gesamtüberleben (OS) festgelegt worden. Die sekundären Endpunkte waren unter anderen das progressionsfreie Überleben (PSA-PFS), das klinische PFS und Nebenwirkungen.
Enzalutamid bewirkte eine deutliche Verlängerung des PSA-PFS und des klinischen PFS. Es traten lediglich 174 PSA-PFS-Ereignisse in der Enza-Gruppe gegenüber 333 Ereignissen in der Standard-Gruppe auf (3-Jahres-Rate an Ereignisfreiheit 67 % bzw. 37 %, HR: 0,39; p < 0,001). Bezüglich klinischen PFS gab es 167 Ereignisse in der Enza- Gruppe und 320 Ereignisse in der Standard-Gruppe (3-Jahres-Rate an Ereignisfreiheit 68 % bzw. 41 %, HR: 0,40; p < 0,001).
Dies übersetzte sich auch auf die mittlere Behandlungsdauer, nach drei Jahren erhielten 62 % der Männer in der Enza-Gruppe und 34 % in der Standard-Gruppe weiterhin das Behandlungsschema. Bei 133 von 201 Patienten (66 %) der Enza-Gruppe und bei 251 von 356 (71 %) der Standard-Gruppe waren Krankheitsprogression oder Tod ursächlich für den Abbruch bzw. das Ende der Behandlung. Das Gesamtüberleben nach drei Jahren lag bei 89 % in der Enzalutamid-Gruppe und 72 % in der Standard-Gruppe.
Erkenntnisreich ist die Betrachtung der Subgruppenanalysen: Bei Patienten, die zusätzlich eine Docetaxel-Chemotherapie erhielten, sowie bei hoher Tumorlast war der zusätzliche Effekt vom Enzalutamid nicht mehr statistisch signifikant ausgeprägt. Somit ist eine Kombination von Docetaxel, Enzalutamid und ADT nicht zu empfehlen.
Die höhere Anzahl an Nebenwirkungen im Zusammenhang mit Enzalutamid stand im Verhältnis zur längeren Behandlungsdauer. Bei der Häufigkeit der Nebenwirkungen pro Personenjahr bestand Ausgewogenheit. Fatigue war in der Enza-Gruppe häufiger. Krampfanfälle traten bei sieben Patienten der Enza- Gruppe (1 %) und bei keinem Patienten der Standard-Gruppe auf. In der Enzalutamid-Gruppe seien Krampfanfälle und andere toxische Wirkungen häufiger gewesen, insbesondere bei jenen, die früh mit Docetaxel behandelt worden waren.2
ARCHES-Studie (Enzalutamid)
Im Rahmen der Phase-III-Studie ARCHES wurden 1.150 Männer mit mHSPC 1 : 1 randomisiert in ADT vs. ADT + Enzalutamid (160 mg/d). Eingeschlossen wurden sowohl High- als auch Low-Volume-Patienten gemäß den CHAARTED-Kriterien sowie solche +/– vorherige Docetaxel-Therapie (18 % der Patienten). Primäres Ziel der Studie war der Nachweis einer Verlängerung des rPFS.3
Bezogen auf den primären Endpunkt zeigte sich in der Gruppe mit ADT + Enzalutamid eine relevante Reduktion des relativen Risikos von 61 % (HR: 0,39; 95%-KI: 0,3–0,5). Ebenso war eine Verlängerung der Zeit bis zum Beginn einer weiteren antineoplastischen Therapie nachweisbar, mit einer Reduktion des relativen Risikos von 72 % in der ADT+Enzalutamid-Gruppe (HR: 0,28; 95%-KI: 0,2–0,4). Im Hinblick auf das Gesamtüberleben zeigt sich bei einem medianen Follow-up von 14,4 Monaten in der ersten Analyse noch kein Unterschied zwischen den Behandlungsarmen (HR: 0,81; 95%-KI: 0,53–1,25). Das Sicherheitsprofil der Kombination aus Enzalutamid und ADT war konsistent mit dem aus früheren Studien bei Männern mit kastrationsresistentem Prostatakarzinom. Unter Enzalutamid zeigte sich entsprechend dem bekannten Nebenwirkungsprofil ein gehäuftes Auftreten von arterieller Hypertonie (8,6 % vs. 6,3 %), Fatigue (24,1 % vs. 19,5 %), kognitiver Beeinträchtigung (4,5 % vs. 2,1 %), Frakturen (6,5 % vs. 4,2 %) und Stürzen (3,7% vs. 2,6%).
Die ARCHES-Studie zeigt zusammengefasst eine klinisch relevante Aktivität von Enzalutamid beim mHSPC in Kombination mit ADT und bestätigt somit die Ergebnisse der ENZAMET-Studie. Die Zulassungserweiterung von Enzalutamid für das mHSPC ist eingereicht.
TITAN-Studie (Apalutamid)
Apalutamid ist ein Inhibitor der Ligandenbindungsdomäne des Androgenrezeptors. Die bisherige Zulassung besteht bei Männern mit einem nichtmetastasierten kastrationsresistenten Prostatakarzinom, die ein hohes Risiko für Metastasen aufweisen (PSA-Verdopplungszeit < 10 Monate).
In die TITAN-Studie wurden 1.052 Patienten mit mHSPC eingeschlossen, definiert als mindestens eine Läsion in der Skelettszintigrafie. Eine vorherige Lokaltherapie sowie eine Systemtherapie mit Docetaxel oder ADT waren erlaubt. 525 Patienten erhielten einmal täglich 240 mg Apalutamid oral, 527 bekamen Placebo. Ergänzend erfolgte in beiden Gruppen eine ADT mit einem LHRH-Analogon. Die Baseline-Charakteristika waren in beiden Armen ausgeglichen: Das Durchschnittsalter betrug 68 Jahre. Insgesamt 16,4 % der Patienten hatten sich einer Prostatektomie unterzogen oder gegen die lokalisierte Erkrankung eine Strahlentherapie erhalten, und 10,7 % hatten zuvor eine Docetaxel-Therapie erhalten. Dabei hatten 62,7 % ein hohes und 37,3 % ein niedriges Metastasenvolumen.4
Bei der ersten Zwischenanalyse nach einer Nachbeobachtung von im Median 22,7 Monaten betrug der Prozentsatz der Patienten mit radiologischem PFS nach 24 Monaten 68,2 % in der Apalutamid- Gruppe und 47,5 % in der Placebo- Gruppe (HR für radiologischen Progress oder Tod 0,48; 95%-KI 0,39–0,60; p < 0,001). Das OS nach 24 Monaten war mit Apalutamid ebenfalls höher als unter Placebo (82,4 % in der Apalutamid- Gruppe vs. 73,5 % in der Placebo-Gruppe; HR für Tod 0,67; 95%-KI 0,51– 0,89; p = 0,005). Nebenwirkungen, die unter Apalutamid/ADT auftraten, waren vor allem Hautausschläge (Apalutamid 27,1 %, Placebo 8,5 %). Auch Hypothyreose, Hypercholesterinämie und Hypertriglyzeridämie traten häufig auf. Die Rate an Therapieabbrüchen aufgrund von Nebenwirkungen lag im Apalutamid- ADT-Arm bei 8,0, verglichen mit 5,3 % im Placebo-ADT-Arm. Nebenwirkungen der Grade 3 und 4 traten in beiden Studienarmen gleich häufig auf (42,2 % vs. 40,8 %).4
Auch beim sekundären Endpunkt, der Zeit bis zum Beginn einer Chemotherapie, bewirkte die Therapie mit Apalutamid eine 61%ige Risikoreduktion gegenüber Placebo. Der Median wurde in beiden Armen noch nicht erreicht (HR 0,39; 95%-KI: 0,27–0,56; p < 0,0001). Ein Wiederanstieg des PSA trat unter Apalutamid deutlich später ein. Die Zeit bis zur PSA-Progression: nicht erreicht vs. 12,9 Monate (HR 0,26; 95%-KI: 0,21–0,32; p < 0,0001). Auch die mediane Zeit von Randomisierung bis Fortschreiten der Erkrankung oder bis zum Tod unter der ersten Folgetherapie (PFS2) war in beiden Armen noch nicht erreicht.
Die Studie bildete die Grundlage für einen Antrag auf Zulassungserweiterung von Apalutamid für das mHSPC bei der U.S. Food and Drug Administration (FDA) sowie der European Medicines Agency (EMA). Die FDA-Zulassung erfolgte Mitte September 2019 und die EU-Zulassung im Jänner 2020.
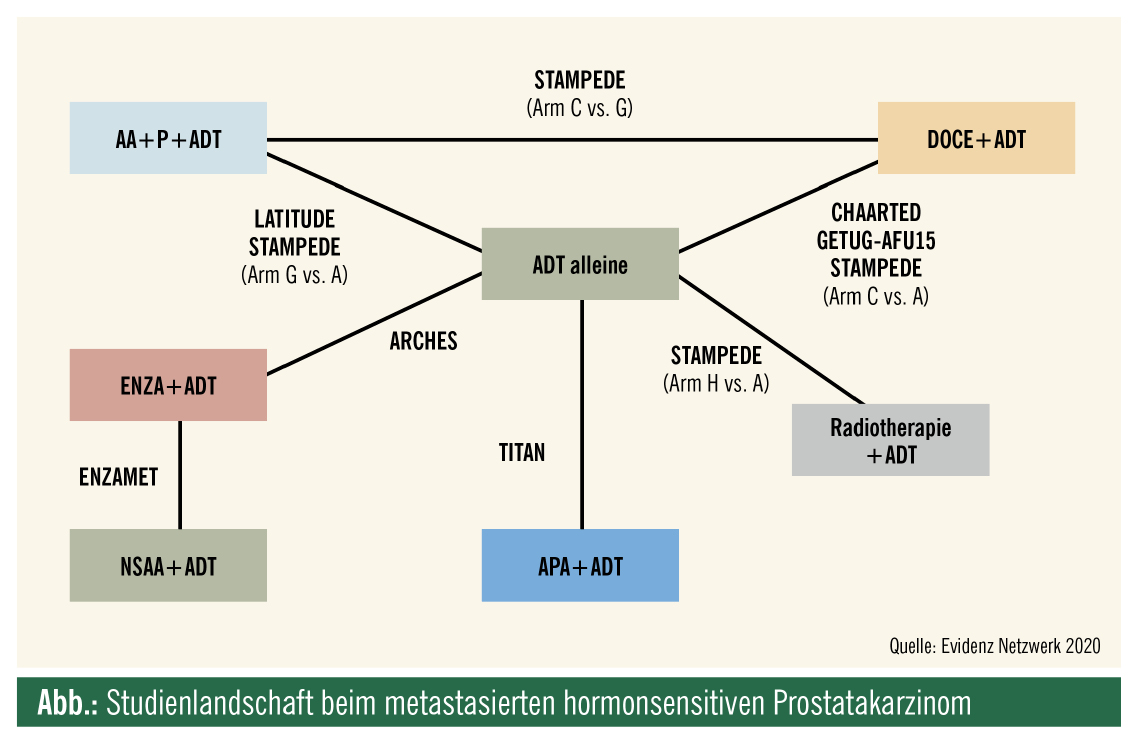
Sequenzierung
Mit den zunehmenden Therapieoptionen beim fortgeschrittenen Prostatakarzinom gewinnt auch die Sequenztherapie an Bedeutung; denn Medikamente, die früh zum Einsatz kommen, sollten die Wirksamkeit von Folgetherapien möglichst nicht beeinträchtigen. Vor diesem Hintergrund ist es interessant, dass in der TITAN-Studie mit dem Endpunkt progressionsfreies Überleben 2 (PFS2) auch die Effektivität der ersten Folgetherapie im mCRPC untersucht wurde.4 Hierbei bestätigte sich, dass deren Wirksamkeit nach einer mHSPC-Therapie mit Apalutamid/ADT erhalten blieb. Ein Grund könnte sein, dass in Biomarker-Analysen die Rate an Androgenrezeptor-Aberrationen und mit ihnen möglicherweise assoziierte Resistenzmechanismen unter Apalutamid plus ADT signifikant geringer war als unter alleiniger ADT. Mit Apalutamid steht ein neuer oraler, antihormoneller Wirkstoff für mHSPC-Patienten zur Verfügung, der unabhängig vom Risiko und von der Vortherapie zum Einsatz kommen kann.
Direkt vergleichende Studien zwischen Abirateron, Apalutamid und Enzalutamid sind derzeit nicht publiziert. In der Patientenaufklärung sollten die Unterschiede der Behandlungsformen detailliert aufgeführt werden und den betroffenen Patienten erläutert werden. Bei der Chemotherapie sind hier die Nebenwirkungen Alopezie, Neutropenie, Fatigue, Nausea und periphere Neuropathie zu nennen, wohingegen bei Abirateron die mineralokortikoiden Nebenwirkungen wie Hypokaliämie, Hypertonie und kardiale Ereignisse im Vordergrund stehen. Bei Apalutamid und Enzalutamid wurden die in den Studien beobachteten Nebenwirkungen oben aufgeführt. Möglichkeiten der Entscheidungshilfe bieten hier auch die Leitlinien der EAU- und S3-Leitlinie, um ein geriatrisches Assessment durchzuführen, um über die Chemotherapiefähigkeit im Einzelfall zu entscheiden.
Biomarker beim mHSPC
Wie beim mCRPC finden sich in ähnlicher Frequenz auch beim mHSPC Defekte in Genen des DNA-Reparaturweges, welche einen prädiktiven Marker für eine Therapie mit PARP-Inhibitoren darstellen können.5 Lässt sich eine Mikrosatelliteninstabilität nachweisen, so eröffnet sich die Möglichkeit einer immunonkologischen Therapie. Die Splicevariante ARV7 zur Vorhersage eines Therapieansprechens auf Androgenrezeptor-Inhibitoren spielt beim mHSPC keine Rolle. Im Hinblick auf prognostische Biomarker ist davon auszugehen, dass ein Verlust von RB1 und TP53, der ja auch mit einer neuroendokrinen Transformation hin zu einem sehr aggressiven Tumor einhergehen kann, auch im mHSPC einen prognostisch ungünstigen Genotyp darstellt.6
1Fizazi K et al., Lancet Oncol 2019; 20:686–700
2Davis ID et al., N Engl J Med 2019; 381:121–131
3Armstrong AJ et al., J Clin Oncol 2019; 37:2974–2986
4Chi KN et al., N Engl J Med 2019; 381:13–24
5Vandekerkhove G et al., Eur Urol 2019; 75:667–675
6Cattrini C et al., Cancers (Basel) 2019; 11

AutorIn: Univ.-Prof. Dr. med. Axel S. Merseburger
Direktor der Klinik für Urologie, Universitätsklinikum Schleswig-Holstein, Campus Lübeck, D

AutorIn: Univ.-Prof. Dr. med. Verena-Wilbeth Sailer
Institut für Pathologie, Universitätsklinikum Schleswig-Holstein, Campus Lübeck, D

Ursprünglich erschienen:
SU 01|2020
SU 01|2020