





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Stammzellen zur Therapie bei Genodermatosen
14. März 2019
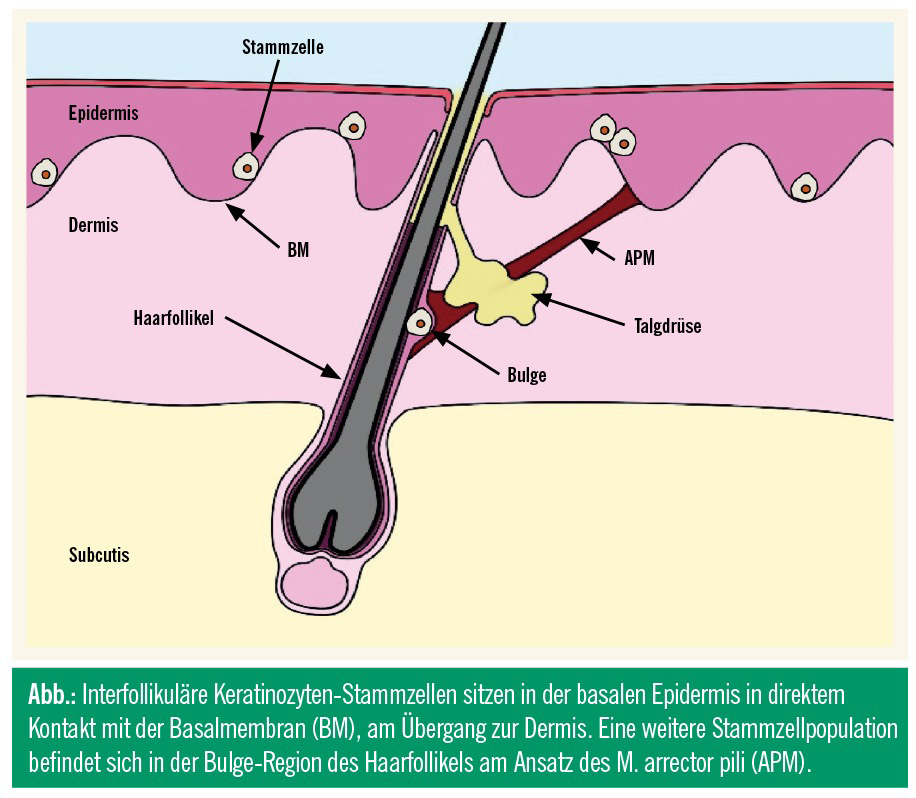
Regeneration durch Stammzellen
Die Epidermis braucht etwa vier Wochen, um sich einmal vollständig zu erneuern. Die Regeneration geht von einer kleinen Zellpopulation von Keratinozyten-Stammzellen aus, die in der untersten Schicht der Epidermis, dem Stratum basale, in Warteposition sitzen und sich nur gelegentlich teilen (Abb.). Bei der Stammzellteilung entstehen entweder Stammzellen, die im Stratum basale bleiben, oder differenzierte Keratinozyten, die sich noch mehrmals teilen können, bevor sie sich von der Basalmembran lösen und in Richtung Hautoberfläche wandern. Dort bilden sie die beinah undurchdringliche Barriere des Stratum corneum, bevor sie schließlich nach und nach abgeschilfert werden. Eine weitere Gruppe von Keratinozyten-Stammzellen befindet sich in der Bulge-Region im oberen Drittel des Haarfollikels am Ansatz des M. arrector pili (Abb.). Diese Zellen sind darauf spezialisiert, Haare zu regenerieren.
Die Regeneration der Epidermis beruht entscheidend auf der Teilungsfähigkeit ihrer Stammzellen. Keratinozyten können aus Biopsien gewonnen und in speziellem Kulturmedium gezüchtet werden. Die Gewinnung und Vermehrung der epidermalen Stammzellen im Labor bei gleichzeitigem Erhalt ihrer Qualität, also der Fähigkeit, sowohl Tochterstammzellen zu bilden als auch sich bei Bedarf zu differenzieren, ist eine Voraussetzung für die Ex-vivo-Gentherapie von Genodermatosen.1
Genodermatosen
Eine wachsende Zahl von Hauterkrankungen kann mittlerweile auf Mutationen im Erbgut zurückgeführt und durch genetische Analyse eindeutig diagnostiziert werden. Bei den meisten dieser Genodermatosen handelt es sich um seltene Erkrankungen, die nach der EURORDIS-Definition weniger als einen von 2.000 Menschen betreffen (https://www.eurordis.org/de/content/was-ist-eine-seltene-krankheit). Auch Epidermolysis bullosa hereditaria (EB) zählt zur Gruppe der seltenen Hauterkrankungen. Sie entsteht durch Mutationen in Genen, die in der Haut exprimiert werden und für die stabile Verbindung von Epidermis und Dermis wichtig sind. Bisher wurden bereits 20 verschiedene Gene identifiziert, die durch Mutationen ihre Funktion verlieren und so die Stabilität der Haut beeinträchtigen können. Bei einigen klinisch diagnostizierten EB-Formen konnte das verantwortliche Gen noch nicht gefunden werden.2
Epidermolysis bullosa
Der Schweregrad der jeweiligen EB-Erkrankung hängt vom betroffenen Gen sowie von Art und Lokalisation der Mutation im Gen ab, aber auch der Erbgang spielt eine Rolle. Bei den schwersten Formen tragen sowohl das paternale als auch das maternale Allel Nonsense-Mutationen, die die Genexpression unterbinden. Beispiele hierfür sind die rezessiv dystrophe EB (RDEB), bei der es durch Mutationen im COL7A1-Gen zum Verlust von Collagen VII in der Basalmembran kommt, und die junktionale EB (JEB). JEB kann durch Mutationen in mehreren Genen ausgelöst werden, wobei Mutationen im LAMB3-Gen, die zum Verlust von Laminin-332 in der Basalmembran führen, am häufigsten sind. Die Haut von betroffenen Patienten zeigt oft schon bei geringer Belastung Blasenbildung. Die Behandlung ist symptomatisch, wobei Wund-management und Schmerztherapie meist im Vordergrund stehen. Bei den schweren progredient verlaufenden EB-Formen können in Bereichen chronischer Wunden sehr aggressive Plattenepithelkarzinome entstehen, die äußerst schwer behandelbar und oft letal sind.
Lange galten Erbkrankheiten wie EB als unheilbar. Mit der Entwicklung moderner molekularbiologischer Methoden und Werkzeuge ist die kausale Gentherapie allerdings in greifbare Nähe gerückt. Es gibt derzeitig zwei Gentherapieansätze für EB: (1) die Ersatz-Gentherapie, die bereits an wenigen Patienten angewendet wurde und derzeit in klinischen Studien getestet wird, und (2) die Designer-Nuklease-vermittelte Gentherapie, die bis jetzt nur an kultivierten Hautzellen von Patienten und vereinzelt im Tierexperiment getestet wurde.3, 4
Gentherapie
Ersatz-Gentherapie bei EB
Die weltweit erste lokale Ersatz-Gentherapie einer erblichen Hauterkrankung wurde bei einem JEB-Patienten mit einer Mutation im LAMB3-Gen durchgeführt und Ende 2006 in der Fachzeitschrift Nature Medicine veröffentlicht.5 Für die Therapie wurde dem Patienten zunächst eine Hautbiopsie entnommen, aus der im Zellkulturlabor Keratinozyten gewonnen wurden. Bei Patienten mit schweren Formen von EB, wie JEB, kann die Zahl der Stammzellen in der Epidermis vor allem in stark betroffenen Arealen deutlich reduziert sein. Dies war auch bei dem ersten Gentherapie-Patienten der Fall, bei dem nur in den schwach betroffenen Handflächen genug Stammzellen vorhanden waren.5 Die aus der Biopsie isolierten Keratinozyten wurden ex vivo mit retroviralen Vektoren transfiziert, die die kodierenden Bereiche (cDNA) eines intakten LAMB3-Gens enthielten. Die LAMB3-cDNA, die von kurzen Stücken des viralen Vektors flankiert ist, wird bei dieser Behandlung nach dem Zufallsprinzip an ganz unterschiedlichen Stellen in die Erbsubstanz eingebaut. Dabei können eine oder auch mehrere cDNAs pro Zelle integrieren. Die Integrationsorte können nicht vorhergesagt werden, sodass nach der Transfektion jede behandelte Zelle ein typisches, einzigartiges Integrationsmuster aufweist, das bei der Zellteilung an ihre Tochterzellen weitergegeben wird.
Die flankierenden viralen Sequenzen sorgen nun dafür, dass die „transgene“ cDNA exprimiert wird und Laminin-332 gebildet werden kann. Die derart modifizierten Keratinozyten können dann vermehrt und im Labor zur Herstellung autologer epidermaler Äquivalente benutzt werden. Die dünnen, durchsichtigen epidermalen Äquivalente können bereits knapp vier Wochen nach der Entnahme der Biopsie transplantiert werden. Da die mutierte Epidermis und der oberste Teil der Dermis vor der Transplantation entfernt werden, regeneriert sich die Epidermis im transplantierten Bereich nur aus transgenen Keratinozyten. Die Transplantate exprimieren LAMB3-cDNA, wodurch Laminin-332 hergestellt werden kann, das dann in der Basalmembran für den Zusammenhalt von Epidermis und Dermis sorgt. Damit die transplantierte Haut dauerhaft Laminin-332 herstellt, muss sie transgene Keratinozyten-Stammzellen enthalten. Nur Keratinozyten-Stammzellen sind in der Lage, sich dauerhaft in der basalen Schicht der Epidermis anzusiedeln, wo sie sich teilen, differenzieren und die darüber liegenden Schichten bilden und theoretisch ein Leben lang regenerieren können.
Die LAMB3-Ersatz-Gentherapie für JEB wurde inzwischen an drei Patienten durchgeführt.5–7 Bei den ersten beiden Patienten wurden nur wenige schwer betroffene, nichtheilende Stellen transplantiert.5, 6 Bei der dritten derartigen Gentherapie wurde erstmals ein Kind behandelt, wobei in insgesamt drei Operationen ca. 80 % der mutierten Epidermis durch transgene autologe Epidermis ersetzt wurden.7 Der durchschlagende Erfolg dieser Therapie, die zu dauerhafter Stabilität der transplantierten Hautbereiche führte, hing wesentlich von der Anwesenheit potenter Keratinozyten-Stammzellen in den Transplantaten ab. Wie nachfolgende Untersuchungen an Hautbiopsien von transplantierten Stellen vier und acht Monate nach dem Eingriff zeigten, regenerierte sich die transgene Haut zunehmend aus den transgenen Stammzellen und ihren Nachkommen. Mit Hilfe der unverwechselbaren LAMB3-cDNA-Integrationsmuster konnte gezeigt werden, dass sich die viral transfizierten Stammzellen aus der ursprünglich entnommenen Biopsie vermehrt hatten und die Epidermis erneuerten, während Keratinozyten, die während der viralen Behandlung bereits differenziert waren, mit der Zeit verschwanden.7
Eine ähnlich aufgebaute Gentherapiestudie, die in den USA an Patienten mit RDEB durchgeführt wurde und bei der die behandelten Stellen auf kleine Bereiche beschränkt blieben, zeigte teilweise Abstoßungsreaktionen.8 Die Ursache dafür ist noch nicht abschließend geklärt. Derzeit laufen in Salzburg zwei weitere Gentherapie-Studien, HOLOGENE17 und HOLOGENE7, an EB-Patienten mit rezessiven Mutationen im COL17A1-Gen (JEB) und im COL7A1-Gen (RDEB).
Während bei rezessiven EB-Erkrankungen mit der Ersatz-Gentherapie die Funktion des mutierten, gewissermaßen stillgelegten Gens durch eine intakte transgene cDNA ersetzt werden kann, ist dies bei dominanten Formen von EB nicht möglich. Dazu zählt EB simplex (EBS), bei der heterozygote Missense-Mutationen in Keratin-Genen (KRT5 oder KRT14) vorliegen. Hier wird das dominante, mutierte Allel exprimiert und fehlerhaftes Keratin gebildet. Um das mutierte Protein, das sich in die Keratinfilamente integriert und das Zytoskelett dadurch schwächt, zu entfernen, muss das mutierte Allel entweder stillgelegt oder repariert werden. Wird das mutierte Allel nicht mehr exprimiert, muss das gesunde Allel die volle Funktion des Gens übernehmen. Mit Designer-Nukleasen ist diese Art des „Gene-Editings“ im Labor bei kultivierten Hautzellen von Patienten bereits möglich.9, 10
Gene-Editing
Designer-Nukleasen wirken wie molekulare Scheren, die mutierte Gene zielgenau ansteuern und schneiden können. Durch den Schnitt im Gen werden die zelleigenen DNA-Reparatursysteme aktiviert, die zur gewünschten Abschaltung oder Reparatur des mutierten Allels führen können. Anders als bei der Ersatz-Gentherapie benötigt man hierfür keine viralen Vektoren, und es kommt daher auch nicht zu zufälligen Integrationen von Transgenen im Genom. Dies ist ein entscheidender Vorteil des Gene-Editings, denn die Integration von cDNA mit flankierenden viralen Bereichen kann zur ungewollten Aktivierung benachbarter Gene führen. Wenn die Expression dieser Gene unkontrollierte Zellteilung nach sich zieht, besteht die Gefahr der Tumorentstehung.
Bei der ersten Generation von Designer-Nukleasen, den Zinkfinger-Nukleasen (ZFN), basiert die Erkennung des gewünschten Gens auf Zinkfinger-Bindedomänen, deren Herstellung schwierig und mit hohen Kosten verbunden ist. Einige Dutzend ZFN wurden und werden mittlerweile in klinischen Gentherapie-Studien getestet. Wegen der aufwendigen Herstellung wurde die zweite Generation von Designer-Nukleasen, TALEN, die nicht nur wesentlich einfacher herzustellen, sondern deren Off-Target-Effekte auch besser abschätzbar sind, gerne angenommen. Trotz ihrer Vorteile gegenüber ZFN haben nur wenige TALEN-basierte Gentherapieansätze den Weg in klinische Gentherapie-Studien geschafft, weil die kostengünstige CRISPR/Cas9-Technologie kurz danach aufkam und durch ihre extrem einfache und kostengünstige Handhabung rasch in den Fokus des experimentellen Gene-Editings rückte. Die rasante Entwicklung auf dem Gebiet bringt ständig neue CRISPR/Cas9-Varianten hervor, mit dem Ziel, die Effizienz und Sicherheit ihrer Anwendung zu verbessern. Die Zahl der registrierten CRISPR/Cas9-vermittelten Gentherapie-Studien steigt, und auch für die Ex-vivo-Korrektur von RDEB-Mutationen in autologen Keratinozyten sind bereits mehrere Gentherapieansätze in der vorklinischen Entwicklung.11–13
Während bei der Ersatz-Gentherapie beinahe jede viral transfizierte Zelle eines oder mehrere Transgene enthält, weisen beim Gene-Editing nicht alle behandelten Zellen die gewünschte Korrektur im mutierten Gen auf, und es können sich zudem Fehler einschleichen. Daher muss im Anschluss an die Behandlung derzeit noch zwingend eine Selektion erfolgen. Unter den korrigierten Keratinozyten müssen darüber hinaus ausreichend Stammzellen vorhanden sein, damit die daraus hergestellten Transplantate in der Lage sind eine dauerhafte Regeneration zu gewährleisten. Der Fokus der Entwicklung liegt daher auf der Steigerung der Effizienz und der Sicherheit der Designer-Nuklease vermittelten Korrektur von Mutationen, so dass die Selektion in der Praxis mit möglichst geringem Aufwand gelingt. Obwohl CRISPR/Cas9 an einem RDEB-Mausmodel bereits mittels Elektroporation direkt an der Haut, also in vivo, getestet wurde4, bedarf es vor einer vergleichbaren Anwendung am Menschen noch einiger Entwicklung. Denn auch hier hängt der langfristige Erfolg der Behandlung von der Effizienz und Sicherheit des Gene-Editings ab und letztendlich von der Korrektur einer ausreichenden Menge epidermaler Stammzellen.
1 Prodinger CM et al., Ann Dermatol 2017; 29(6):667–87
2 Has C et al., Exp Dermatol 2018; DOI: 10.1111/exd.13668 (Epub ahead of print)
3 De Rosa LK et al., Expert Opin Orphan Drugs 2018; 6(4):283–93
4 Wu W et al., Proc Natl Acad Sci U S A 2017; 114(7):1660–5
5 Mavilio F et al., Nat Med 2006; 12(12):1397–402
6 Bauer JW et al., J Invest Dermatol 2017; 137(3):778–81
7 Hirsch T et al., Nature 2017; 551(7680):327–32
8 Siprashvili Z et al., JAMA 2016; 316(17):1808–17
9 Aushev M et al., Mol Ther Methods Clin Dev 2017; 6:112–23
10 Kocher T et al., Mol Ther 2017; 25(11):2585–98
11 Hainzl S et al., Mol Ther 2017; 25(11):2573–84
12 Izmiryan A et al., Mol Ther Nucleic Acids 2018; 12:554–67
13 Osborn MJ et al., Mol Ther 2013; 21(6):1151–9


AutorIn: Dr. habil. Julia Reichelt
Leiterin der EB-Forschung am EB-Haus Austria in Salzburg, Universitätsklinik für Dermatologie, Paracelsus Medizinische Privatuniversität Salzburg

Ursprünglich erschienen:
SD 01|2019
SD 01|2019
Herausgeber: Ao. Univ.-Prof. Dr. Christoph Höller, Assoc. Prof. Priv.-Doz. Dr. Constanze Jonak, Univ.-Prof. Dr. Rainer Kunstfeld, Univ.-Prof. Dr. Hubert Pehamberger
Publikationsdatum: 2019-03-14
Zur Ausgabe »
Publikationsdatum: 2019-03-14
Zur Ausgabe »
