





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Alpha-1-Antitrypsinmangel
24. Mai 2019
Der Alpha-1-Antitrypsinmangel ist eine vererbbare Erkrankung, welche erstmals 1963 von C. B. Laurell und S. Eriksson1 beschrieben wurde und zu einer der häufigsten genetischen Ursachen für Lungen- und Lebererkrankungen zählt.2–4
Pathophysiologie
Alpha-1-Antitrypsin (AAT) ist ein Akute-Phase-Protein, welches im Rahmen von Infektionen vermehrt gebildet wird und als Proteinaseinhibitor eine gewebeschützende Funktion einnimmt: Es neutralisiert die bei einer Inflammation freigesetzten proteolytischen Enzyme, allen voran die von neutrophilen Granulozyten ausgeschüttete Elastase. Diese übernimmt im Rahmen einer Entzündungsreaktion im Bereich der Atemwege die Aufgabe, bakterielle Proteine durch einen proteolytischen Abbau zu zersetzen.
Bei einem Mangel an AAT kommt es zu einer Dysbalance im Zusammenspiel von Proteinasen und Antiproteinasen, und es wird gesundes Lungengewebe geschädigt. Als langfristige Folge entsteht ein (meist panazinäres) Emphysem.4, 5
Genetik
AAT wird durch ein einziges Gen (SERPINA1) auf Chromosom 14 codiert. Die reguläre AAT-Form wird als Proteinaseinhibitor M (PiM) deklariert. Bei ca. 95 % der Bevölkerung in Mitteleuropa liegt der Genotyp PiMM und somit die Produktion normaler AAT-Mengen vor. Der normale Serumspiegel liegt zwischen 90 und 200 mg/dl.2, 6
Durch eine Punktmutation im SERPINA1-Gen kommt es zu einer Fehlfaltung der Proteine im endoplasmatischen Retikulum der Hepatozyten mit Polymerbildung. Die AAT-Moleküle können in dieser Form nicht sezerniert werden (ein erniedrigter Serumspiegel ist messbar) und werden in den Leberzellen abgelagert. Dies kann eine Hepatopathie auslösen. Diesen pathologischen Mechanismus findet man bei Z-Allelen (PiZ) und S-Allelen (PiS).6, 7
Es kann aber durch die Mutation auch zu einer Funktionsstörung des AAT-Allels kommen (PiF) oder durch ein Stoppcodon zu einer fehlenden Proteinbildung (PiNull).2, 4, 6
Bislang sind über 100 genetische Varianten des AAT-Proteins bekannt. Der AAT-Mangel wird autosomal kodominant vererbt.
Einen relevanten Krankheitswert findet man bei homozygoten und compound-heterozygoten Formen. Am häufigsten wird ein klinisch relevanter Mangelzustand durch das homozygote Vorkommen der Z-Mutation verursacht (PiZZ). Bei der compound-heterozygoten Form PiSZ kommt es z. B. zu einem Zusammentreffen zweier verschiedener und jeweils für sich pathologischer Allele, das Erkrankungsrisiko wird als mittelgradig eingestuft.
Häufig liegt bei gescreenten Patienten die heterozygote Form PiMZ vor. Der Patient ist somit Träger der Mangelmutation, hat selbst aber nur ein geringes Risiko zu erkranken.7–10
Prävalenz
Die Prävalenz des AAT-Mangels in der westeuropäischen Bevölkerung beträgt in etwa 1 : 2.500.12 In Europa lassen sich topografische Unterschiede bei den Mutationsvarianten erheben: So wird die höchste Prävalenz der PiZ-Mutationen in nordwesteuropäischen Ländern, die der PiS-Formen im südlichen Europa mit Gipfel auf der iberischen Halbinsel dokumentiert.4, 6
Der Genotyp PiZZ liegt in 95 % der betroffenen Patienten vor. Daten aus Österreich zeigen, dass die Prävalenz für diesen Genotyp bei ca. 1.500 liegen muss. Im nationalen Register (Austrian Alpha-1 Lung Registry) sind bislang nur knapp 280 Patienten mit Genotyp PiZZ erfasst.4, 10–12
Klinik
Die klinische Ausprägung der Erkrankung ist abhängig vom Genotyp und dem Serumspiegel. Sie reicht von Symptomfreiheit bis zu schweren Lungen- und Lebererkrankungen. Eine pulmonale Symptomatik beginnt meist unspezifisch mit Dyspnoe, Husten und Auswurf und kann bis zur Entwicklung eines schweren Lungenemphysems fortschreiten. Im Vergleich zur COPD ist der Erkrankungsbeginn typischerweise früher, meist bereits ab dem 30.–35. Lebensjahr. Wegen der geringen Spezifität der Beschwerden wird die Diagnose erst nach durchschnittlich 4–8 Jahren gestellt.2, 6, 8, 13
Bei hepatischer Beteiligung können bereits im Neugeborenenalter ein prolongierter Ikterus oder eine Erhöhung der Lebertransaminasen bis hin zum klinischen Bild einer Hepatitis auftreten. Im Verlauf ist sowohl eine vollständige Ausheilung als auch die frühe Entstehung einer Leberzirrhose möglich. In der Literatur beschrieben wird auch, dass Patienten, welche in der Kindheit keine Leberauffälligkeiten zeigten, mit einem AAT-Mangel Genotyp PiZZ im Erwachsenenalter ein erhöhtes Risiko für die Entwicklung einer Leberzirrhose und eines hepatozellulären Karzinoms haben.6
Prinzipiell kann gleichzeitig eine Leber- und Lungenbeteiligung bei AAT-Mangel auftreten, das Vollbild der Erkrankung mit sowohl schwerem Lungenemphysem als auch Leberzirrhose ist allerdings selten.
Assoziiert mit einem AAT-Mangel kann in seltenen Fällen auch eine nekrotisierende Panniculitis und sekundäre Vaskulitis sein.4
Diagnostik
Der AAT-Mangel wird bei weitem zu selten diagnostiziert. Grund ist die hohe Variabilität in der Symptomausprägung: Diese reicht von respiratorischen Symptomen, ähnlich der COPD und des Asthma bronchiale, bis zu gänzlich asymptomatischen Verläufen. Wie in der Leitlinie der ATS/ERS7 empfohlen, sollte jeder Patient mit COPD – ungeachtet des Alters – auf einen AAT-Mangel getestet werden. Dies gilt ebenso für Patienten mit einer kryptogenen Lebererkrankung, nekrotisierenden Pannikulitis und Granulomatose mit Polyangiitis.
Bei gesicherter Diagnose wird ein Screening der Familienmitglieder empfohlen.
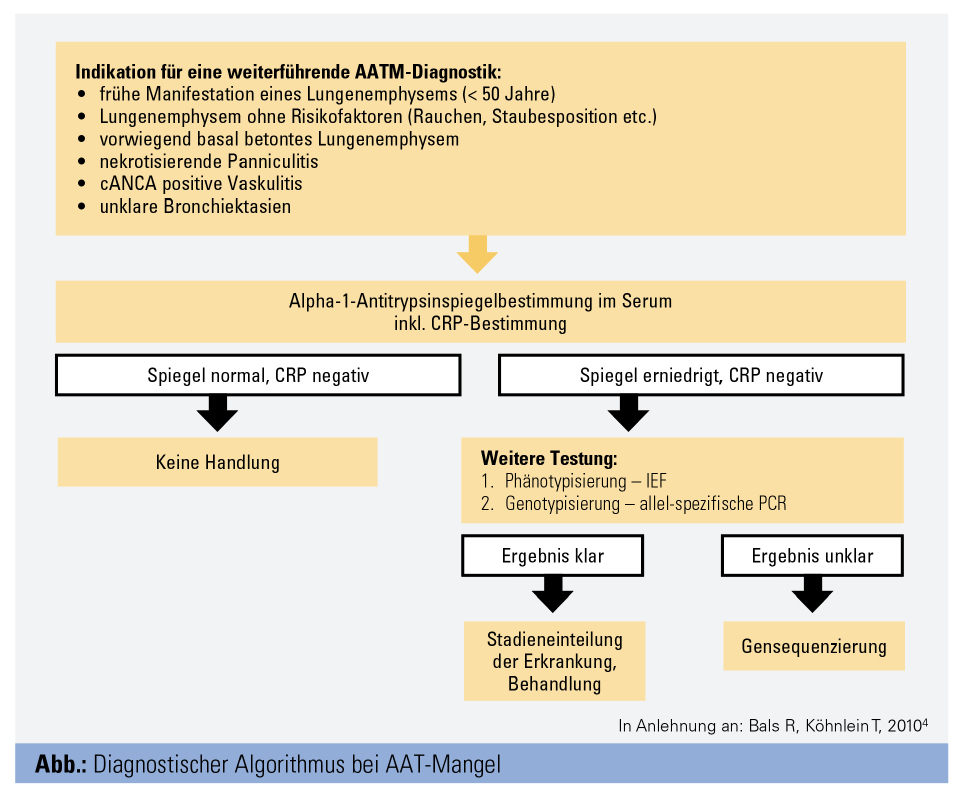
Zur Testung gehört initial die Bestimmung des Serumspiegels. Da es sich bei AAT um ein Akute-Phase-Protein handelt, kann der Serumspiegel bei Entzündungen erhöht sein. Es soll daher gleichzeitig eine Bestimmung des CRP erfolgen. Bei erniedrigtem oder im unteren Normbereich liegendem Serumspiegel von AAT müssen weitere genetische Untersuchungen eingeleitet werden: die Phänotypisierung mittels isoelektrischer Fokussierung (IEF) und Genotypisierung mittels PCR. Bei Verdacht auf eine seltene Mutation, d. h. bei unschlüssigen Befundkonstellationen, ist eine Gensequenzierung notwendig (Abb.).7, 8, 10
Nach Diagnosesicherung erfolgt eine umfassende erste klinische, funktionelle und radiologische Evaluierung.3
Therapie
Die Behandlung des AAT-Mangels mit Lungenbeteiligung folgt einem multimodalen Konzept, welches sich im Wesentlichen nicht von den Empfehlungen für COPD unterscheidet.14 Unumgänglich ist der komplette Rauchstopp bzw. das Schaffen einer rauch- und staubfreien Umgebung. Dazu gehört ggf. auch eine berufliche Umorientierung.3, 7
Ist eine Lungenfunktionseinschränkung bereits manifestiert, kann bei klinisch relevanten Mangelvarianten (PiZZ, PiSZ, PiZ0, Pi00) eine Augmentationstherapie eingeleitet werden. Diese besteht in einer einmal wöchentlichen intravenösen Gabe von hochgereinigtem AAT. Ziel dabei ist es, den Serumspiegel über die „protektive Schwelle“ (> 80 mg/dl) anzuheben, um eine fortschreitende Lungenzerstörung hintanzustellen.15, 16
Die Indikation zur Therapie muss durch ein Kompetenzzentrum für AAT gestellt werden. Hier erfolgt auch die Aufnahme der Patienten in das nationale Register.
Sind alle Therapiemöglichkeiten ausgeschöpft, wird bei geeigneten Patienten eine Lungentransplantation angestrebt.7
Resümee
Der AAT-Mangel ist eine unterdiagnostizierte Erkrankung. Ein flächendeckendes Screening von allen COPD-Patienten führt zu einer früheren Diagnose, was wiederum den Krankheitsverlauf deutlich positiv beeinflusst.
Eine lebenslange Augmentationstherapie kann für Patienten mit manifester pulmonaler Erkrankung angeboten werden.
1 Laurell CB et al., J Clin Lab Invest 1963; 15:132
2 Silvermann EK et al., N Engl J Med 2009; 360(26):2749–57
3 Sandhaus RA et al., Chronic Obstr Pulm Dis 2016; 3:668–82
4 Bals R, Köhnlein T. (Hrsg.), Alpha-1-Antitrypsin-Mangel: Pathophysiologie, Diag
nose und Therapie 2010. Thieme; 1. Aufl.
nose und Therapie 2010. Thieme; 1. Aufl.
5 Gadek J et al., J Clin Invest 1981; 68:889–98
6 Janciauskiene SM et al., Respir Med 2011; 105:1129–39
7 American Thoracic Society/European Respiratory Society., Am J Respir Crit Care Med 2003; 168:818–900
8 Schroth S et al., Pneumologie 2009; 63(6):335–45
9 Zorzetto M et al., Clin Chem 2008; 54(8):1331–8
10 Luisetti M, Breathe 2007; 4:39–46
11 Blanco I et al., Eur Respir J 2006; 27:77–84
12 https://www.orpha.net/consor/cgi-bin/OC_Expphp?Expert=60&lng=DE, letzte Aktualisierung des Artikels: Juni 2008, abgerufen am 20. 04. 2019
13 Campos MA et al., Chest 2005; 128:1179–86
14 Vogelmeier C et al., Pneumologie 2018; 72:253–308
15 Turino GM et al., Am J Resp Crit Care Med 1996; 154:1718–25
16 Köhnlein T et al., Pneumologie 2014; 68:492–5

AutorIn: Dr. Eva Traunmüller-Wurm
Abteilung für Lungenkrankheiten, Klinikum Wels-Grieskirchen

AutorIn: Dr. Kristina Kutics

Ursprünglich erschienen:
UIM 04|2019 Themenheft Orphan Diseases
UIM 04|2019 Themenheft Orphan Diseases