





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Pulmonale Vaskulitis – Histopathologie und morphologische Veränderungen
11. November 2011
Histopathologisch besteht eine akute oder chronische zelluläre Inflammation innerhalb der Gefäßwand, diese führt in der Regel zur Destruktion des Blutgefäßes und umgebender Lungengewebsnekrose. Gewöhnlich ist dieser Prozess mit einer systemischen Vaskulitis verbunden, kann aber auch isoliert auftreten.
Die an der Lunge morphologisch sichtbaren Veränderungen sind sehr variabel und leider nicht spezifisch ausgeprägt. Sie reichen von diffusen oder fokalen milchglasartigen Verschattungen bis zu kavernösen Noduli. Auch diffuse alveoläre Hämorrhagien können auftreten, ebenso wie direkte Veränderungen an den Gefäßen selbst (z.B. Gefäßstenosen, Gefäßverschlüsse) Die Einteilung erfolgt nach der Chapel-Hill-Klassifikation, die alle entzündlichen Gefäßerkrankungen umfasst. Sie ist jedoch im Falle der pulmonalen Vaskulitis eine modifizierte Klassifikation (Tab. 1), da die in der Chapel-Hill-Klassifikation erfassten Gefäßentzündungen selten eine pulmonale Beteiligung aufweisen (z.B. Riesenzellarteriitis als typisches Beispiel), die nicht inkludierten (Tab. 1) jedoch in sehr hohem Ausmaß.
| Tab.: Modifizierte Chapel-Hill-Klassifikation der pulmonalen Vaskulitis | |
|---|---|
| In der Chapel-Hill-Nomenklatur inkludiert | In der Chapel-Hill-Nomenklatur nicht inkludiert |
|
|
| Modifiziert: 1) Riesenzell-Arteriitis (GCA), Polyarteriitis nodosa (PAN) und Kawasaki-Syndrom kaum pulmonale Beteiligung 2) nicht inkludiert, jedoch hohe pulmonale BeteiligungProblem: die Klassifikation nach Gefäßgröße |
|
| Chung M.P. et al., Radiology 2010; 255:322-41 | |
Klinik
Die Verdachtsdiagnose der pulmonalen Beteiligung ergibt sich aus der klinischen Präsentation des Patienten.
In der Regel ist dies eine Bandbreite an multisystematischen Manifestationen wie
-
diffuse alveoläre Hämorrhagie (Trias: diffuse alveoläre Infiltration, Hämopytsen, Abfall HK, Hb)
-
akute Glomerulonephritis
-
Pulmonary-Renal Syndrome (DAH + GN)
-
Läsionen im oberen Respirationstrakt
-
Noduli/kavernöse Läsionen (Bildgebung)
-
Mononeuritis multiplex
-
Multisystembefall
-
palpable Purpura
Typische Laborparameter
Anti-neutrophile zytoplasmatische Antikörper (ANCA) sind, wenn sie positiv sind und zum Gesamtkrankheitsbild passen, auch ein Parameter für eine pulmonale Vaskulitis im Rahmen der Systemerkrankung, jedoch für eine Verlaufsdiagnostik sind sie nicht geeignet (Ausschluss Relaps).
Beispiele einer pulmonalen Vaskulitis im Rahmen einer Systemerkrankung
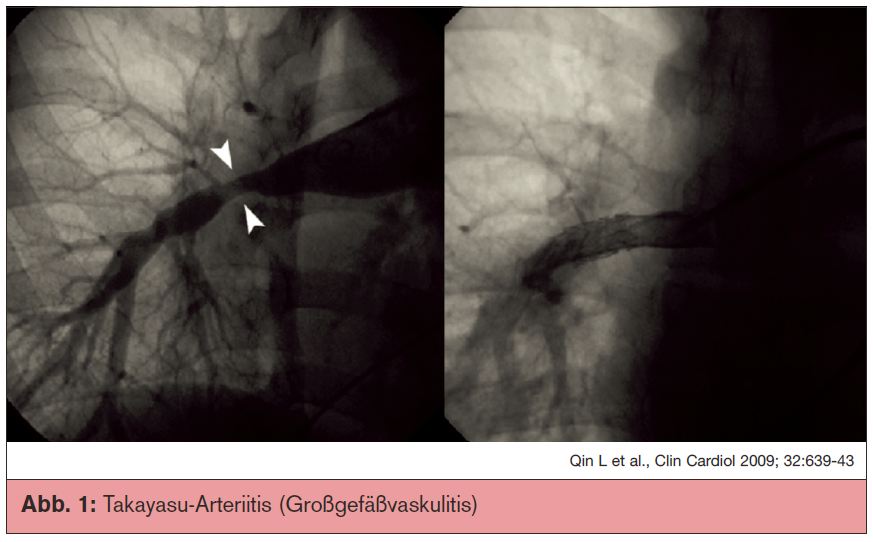
Takayasu-Arteriitis (Großgefäßvaskulitis): Der Morbus Takayasu ist eine chronisch inflammatorische Arteriitis mit Befall der großen Gefäße, hauptsächlich der Aorta und ihrer abgehenden Äste. Sie führt zu fortschreitender Gefäßwandfibrose und Lumeneinengung sowie gelegentlich zur Zerstörung der Adventitia mit konsekutiver Aneurysmabildung vor allem im Bereich der elastischen Arterien. Das Typische an dieser Erkrankung ist, dass vor allem junge Frauen vor dem 40. Lebensjahr betroffen sind. Die Inzidenz ist selten (0,12 bis 0,26/100.000), die Inzidenz der pulmonalen Beteiligung mit 15% vergleichsweise hoch. Die Klinik ist unspezifisch mit Allgemeinsymptomen wie Müdigkeit, Mattigkeit und Abgeschlagenheit sowie Gewichtsverlust, Nachtschweiß und Fieber. Abb. 1 zeigt ein Angiogramm mit einer Takayasu-Arteriitis und Beteiligung der Pulmonalgefäße mit Stenosen und nachgeschalteten Aussackungen der Arterien.
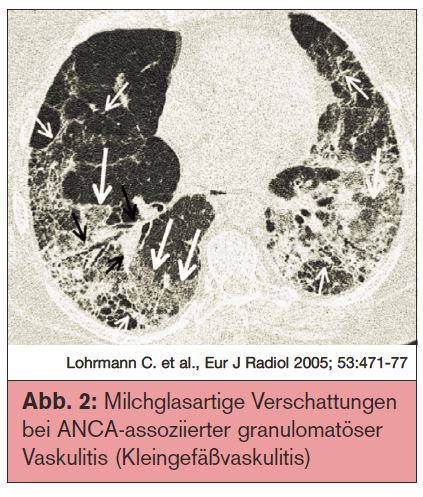
ANCA-assoziierte granulomatöse Vaskulitis (Kleingefäßvaskulitis): Die Klinik spiegelt eine typische Trias wider:
-
oberer Respirationstrakt: Sinusitis, Otitis, Ulzerationen, Knochendeformationen, subglottische oder Bronchialstenosen
-
unterer Respirationstrakt: Husten, Thoraxschmerz, Dyspnoe, Hämoptysen
-
Glomerulonephritis
Das mediane Befallsalter liegt bei 45 Jahren mit einer Inzidenz von 1/100.000. Lebensbedrohend ist vor allem der pulmonale Befall mit pulmonalen Hämorrhagien, die immer eine Notfallsituation darstellen. Typische Befunde einer pulmonalen Beteiligung sind Noduli oder Konsolidierungsareale mit Kavernierung sowie eine Bronchialwandverdickung, bei 70% der Patienten im Bereich der großen Bronchien, bei 30% ist die subglottische Trachea die Prädilektionsstelle. In 25-50% der Patienten findet man diffuse milchglasartige Verschattungen im Thoraxröntgen, die eine diffuse alveoläre Hämorrhagie repräsentieren, verursacht durch eine nekrotisierende Kapillaritis (Abb. 2).
Morbus Behcet: Eine Vaskulitis, die nicht in der Chapel-Hill-Klassifikation vertreten ist, die aber eine sehr hohe und wenn vor allem fatale Beteiligung der Pulmonalgefäße darstellt, ist der Morbus Behcet.
Es handelt sich um eine chronische systemische Vaskulitis, die vor allem bei jungen Männern um das 30. Lebensjahr auftritt, die aus dem asiatischen Raum stammen (ehemalige Seidenstraße). In 8% besteht eine pulmonale Beteiligung, die vor allem in der Form eines Pulmonalarterienaneurysmas auftritt.
Im befallenen Gefäß kommt es zur Mediaverdickung, zum Splitting der elastischen Fasern, zur perivaskulären Zellinfiltration mit nachfolgendem Gefäßverschluss entweder im arteriellen oder venösen Schenkel.
Die “maligne Form” des Morbus Behcet mit einer pulmonalen Beteiligung ist der Morbus Hughes Stovin. Diese sehr schwere Verlaufsform des Morbus Behcet betrifft vor allem junge Männer zwischen dem 20. und 40. Lebensjahr und ist gekennzeichnet durch eine spezifische Trias aus systemischen Thromboembolien, Pulmonalarterienaneurysmata und pulmonalen Hämorrhagien. Die Letalitätsrate liegt bei 80% (Abb. 3).

Kollagenosen: Die derzeit bekannteste Manifestation einer “pulmonalen Vaskulitis” besteht im Zusammenhang mit Kollagenosen. Hier ist sicher die häufigste Manifestation die Sklerodermie und die damit auftretenden pulmonalen Affektionen in Form einer PAH, aber auch systemischer Lupus erythematodes und andere Kollagenosen weisen eine Lungenbeteiligung auf.
Therapie
Die Therapie richtet sich generell nach der zugrunde liegenden Grunderkrankung und reicht von der systemischen Verabreichung von Glukokortikoiden bis zur ausgeprägt immunsuppressiven Therapie.
Zusammenfassung: Das Auftreten einer pulmonalen Vaskulitis ist häufiger als oft bedacht. Bei jeglicher Autoimmunerkrankung kann eine solche auftreten, daher sollte auf augenscheinliche pulmonale Symptome geachtet werden. Das Problem der Diagnostik liegt im bunten Erscheinungsbild, die Problematik in der oft schweren und auch tödlichen Verlaufsform der Erkrankung.

Ursprünglich erschienen:
UIM 09|2011
UIM 09|2011
Herausgeber: Univ.-Prof. Dr. Günter J. Krejs, Österreichische Gesellschaft für Innere Medizin
Publikationsdatum: 2011-11-11
Zur Ausgabe »
Publikationsdatum: 2011-11-11
Zur Ausgabe »