




















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Bessere Aussichten bei Lupus
21. Februar 2019
Unter dem Begriff des systemischen Lupus erythematodes (SLE) wird ein heterogener Symptomenkomplex, der mit Autoimmunität gegen Zellkernbestandteile einhergeht, zusammengefasst. Die Heterogenität der Symptome und Verlaufsformen erschwert nicht nur die Diagnose und Therapie des SLE, sie hemmt auch die Entwicklung dringend benötigter neuer Therapieansätze.
Aktuelle Therapien und deren Risiken
Der SLE ist eine potenziell lebensbedrohliche Erkrankung. Unbehandelt starben 40 % der Betroffenen innerhalb von drei Jahren nach Diagnosestellung. Diese erschreckend hohe Mortalität, wie sie noch in den 1940er Jahren berichtet wurde1, konnte vor allem durch die Entdeckung der Kortikosteroide und die Verfügbarkeit der Nierenersatztherapie deutlich verringert werden. Dennoch ist der SLE auch heute noch eine der häufigsten Todesursachen junger Frauen in den USA, und SLE-Patienten weisen eine 5-fach erhöhte Sterblichkeitsrate im Vergleich zur übrigen Bevölkerung auf.2
Neben der Erkrankung selbst tragen hierzu auch Komplikationen der immunsuppressiven Therapie bei. Insbesondere Kortikosteroide sind mit erhöhter Mortalität durch Infektionen, kardiovaskulären Ereignissen und osteoporotischen Frakturen vergesellschaftet.3 Dauer und Dosis sind die wesentlichen Faktoren für das Auftreten von kortikosteroidinduzierten Nebenwirkungen. Aus diesem Grund sollten Kortikosteroide nur für die kurzzeitige, überbrückende Therapie von Krankheitsschüben eingesetzt werden, während die oft notwendige Langzeittherapie durch immunmodulatorische Medikamente erfolgen soll (Tab.).
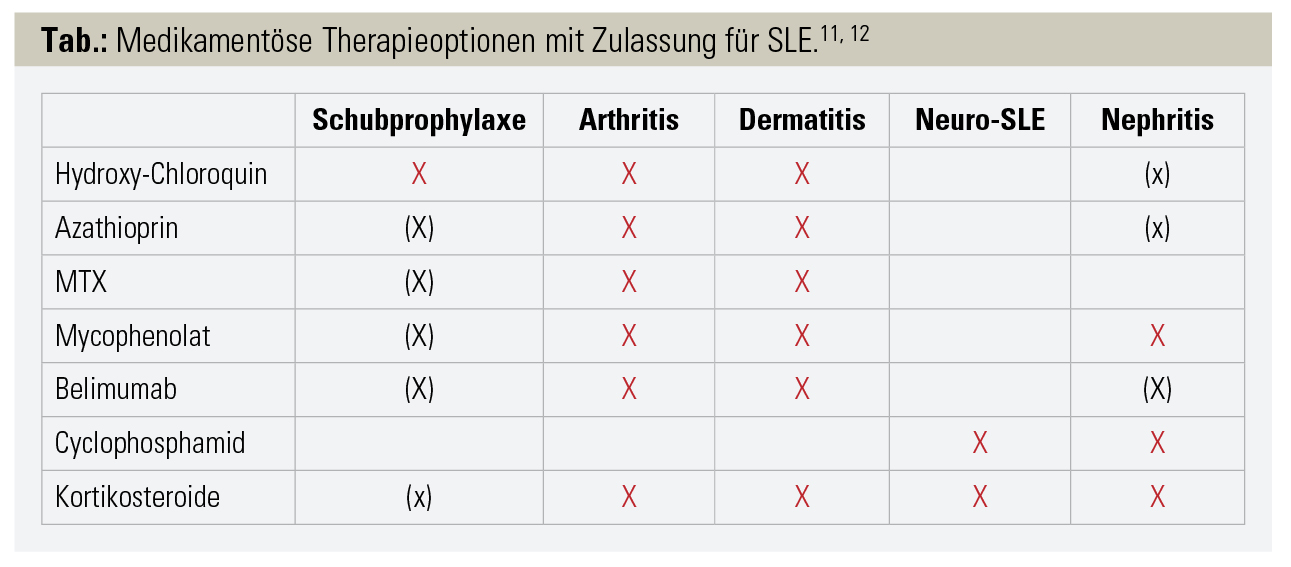
Insbesondere (Hydroxy-)Chloroquin, das auch als Antimalariamedikament verwendet wird, kommt hierbei wesentliche Bedeutung zu. Hydroxychloroquin vermindert die Anzahl an Erkrankungsschüben, verbessert das Überleben und vermindert die Wahrscheinlichkeit von SLE-bedingten Insulten und Thrombosen.4 Demgegenüber steht die Gefahr der Nebenwirkung einer irreversiblen Makulopathie, welche in Extremfällen bis zur Erblindung führen kann. Erste Anzeichen dieser Makulopathie können heute mittels moderner optischer Kohärenztomografie und Computerperimetrie frühzeitig erkannt werden, sodass Visusbeeinträchtigungen durch das rechtzeitige Absetzen des Medikamentes verhindert werden können. Neuere Ergebnisse zeigen nun, dass die Makulopathie streng dosis- und zeitabhängig ist.5 Erste Anzeichen der Makulopathie treten frühestens nach 5 Therapiejahren auf; in der gebräuchlichen Dosierung von 5–6 mg/kg Körpergewicht sogar erst nach 10 Therapiejahren. Aus diesem Grund hat sich die Amerikanische Gesellschaft der Ophthalmologen auf die Richtlinie geeinigt, dass Patienten unter Hydroxychloroquintherapie zu Beginn der Therapie und nach 5 Therapiejahren jährlich eine optische Kohärenztomografie und Computerperimetrie erhalten sollen.6 Unter diesen Vorkehrungen kann das für die SLE-Therapie so wertvolle Medikament weitgehend sicher angewendet werden.
Neue Therapieansätze
Im Gegensatz zur beständigen Innovation in der Therapie anderer Autoimmunerkrankungen, wie der rheumatoiden Arthritis, der Psoriasis vulgaris oder den chronisch entzündlichen Darmerkrankungen, kommt die Entwicklung neuer Medikamente zur Therapie des SLE nur stockend voran (Abb.). So wurde in den letzten 30 Jahren lediglich ein einziges neues Medikament, Belimumab, für den SLE zugelassen. Dies liegt jedoch nicht an mangelndem wissenschaftlichem Interesse für den SLE; mehr als 150 klinisch therapeutische SLE-Studien wurden in den letzten Jahrzenten durchgeführt. Weshalb konnten so viele dieser Studien ihren primären Endpunkt nicht erreichen? Zum einen ist der SLE eine heterogene Erkrankung, welche sich daher per se nur schlecht für therapeutische Studien eignet. So wird eine Studie, in der SLE-Patienten mit Hautbeteiligung und Arthralgie in gleicher Weise behandelt werden wie Patienten mit lebensbedrohlicher Nieren- oder ZNS-Beteiligung, nur selten den gemeinsamen Endpunkt erreichen. Auch worin dieser Endpunkt bestehen soll, was also ein klinisch sinnvolles Therapieziel ist, war bisher umstritten. Aktuelle Bemühungen zur Definition von Remission und niedriger Krankheitsaktivität sowie die neuen Klassifikationskriterien des SLE lassen jedoch zukünftig auf effizientere Studien hoffen.7 Zum anderen ist unser Wissen zur Pathophysiologie der Erkrankung, trotz großer Fortschritte in den letzten Jahren, nach wie vor beschränkt. Dies erschwert die Identifikation von lohnenden therapeutischen Eingriffsmöglichkeiten auf molekularer und zellulärerer Ebene.
Auf bessere Aussichten in der Therapie des SLE lassen dennoch einige erfolgreich abgeschlossene Phase-II-Studien hoffen: So zeigte der Interleukin-12/23-Hemmer Ustekinumab, der unter anderem bereits für die Behandlung der Psoriasis vulgaris und des Morbus Crohn zugelassen ist, in einer rezenten Studie ein signifikantes Ansprechen bei 60 % der untersuchten SLE-Patienten.8 Auch die Rate an Krankheitsschüben konnte gegenüber Placebo deutlich verringert werden. Der Januskinasehemmer Baricitinib zeigte in einer anderen Phase-II-Studie einen positiven Einfluss auf Haut- und Gelenkbeteiligung sowie auf das Auftreten von Krankheitsschüben.9 Sirolimus, ein mTOR-Inhibitor, der in der Transplantationsmedizin erfolgreich eingesetzt wird, führte zu einer Reduktion der klinischen und immunologischen Krankheitsaktivität von SLE-Patienten.10
Resümee
Die Behandlung von Patienten mit SLE kann eine Herausforderung darstellen und muss an die individuellen Krankheitssymptome angepasst werden. Therapieassoziierte Risiken sollen, zum Beispiel durch möglichst kurze Kortikosteroidexposition, vermieden werden. Verbesserte Studiendesigns und neue Erkenntnisse in der Pathophysiologie der Erkrankung lassen auf bessere Aussichten bei Lupus hoffen.
1 Jessar RA et al., Ann Intern Med 1953; 38(4):717–31
2 Singh RR et al., Lupus 2018; 27(10):1577–81
3 Apostolopoulos D et al., Rheumatology (Oxford) 2017; 56(1):i114–i122
4 Tang C et al., Intern Med J 2012; 42(9):968–78
5 Melles RB et al., JAMA Ophthalmol 2014; 132(12):1453–60
6 Marmor MF et al., Ophthalmology 2016; 123(6):1386–94
7 Morand EF et al., Best Pract Res Clin Rheumatol 2017; 31(3):342–50
8 van Vollenhoven RF et al., Lancet 2018; 392(10155):1330–9
9 Wallace DJ et al., Lancet 2018; 392(10143):222–31
10 Lai ZW et al., Lancet 2018; 391(10126):1186–96
11 Bertsias GK et al., Ann Rheum Dis 2012; 71(11):1771–82
12 Bertsias G et al., Ann Rheum Dis 2008; 67(2):195–205



AutorIn: Assoz. Prof. Priv.-Doz. Dr. Martin H. Stradner
Klinische Abteilung für Rheumatologie und Immunologie,Universitätsklinik für Innere Medizin, Medizinische Universität Graz

Ursprünglich erschienen:
UIM 01|2019 Themenheft Rheumatologie
UIM 01|2019 Themenheft Rheumatologie