


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Philadelphia-negative myeloproliferative Neoplasien
11. November 2011
Gleich zu Beginn des Kongresses widmete sich ein Satellitensymposium den relevantesten klinischen Neuerungen auf dem Gebiet der MPN. Wie von Professor Petro Petrides von der Ludwig-Maximilians Universität München zusammengefasst wurde, dürften nicht nur ältere und mittlerweile bei MPN eigentlich obsolete Zytoreduktiva (z.B. Radiophosphor 32P, Melphalan, Busulphan, Chlorambucil) mit einem relevanten Risiko einer leukämischen Transformation behaftet sein. Auch eine Langzeittherapie mit dem heute in der Routine eingesetzten Hydroxyurea wird immer wieder mit einer deutlichen Steigerung der Leukämierate in Zusammenhang gebracht. Genomische und posttranslationelle Einflüsse auf den Tumorsuppressor p53 scheinen hierbei eine zentrale Rolle zu spielen; sowohl Deletionen seines Genlocus, 17p, als auch eine direkte Hemmung der Transaktivierung des p53-Moleküls werden als mögliche Mechanismen diskutiert. Wie Petrides betonte, besitzt diese mögliche Leukämogenität klinische Relevanz: In einer prospektiven französischen Langzeitstudie (Kiladjian J. J., J Clin Oncol 2011; 29:3907) lag die kumulative Inzidenz einer leukämischen Transformation (in akute myeloische Leukämien und myelodysplastische Syndrome) unter Polycythaemia-vera-Patienten, die langfristig mit Hydroxyurea behandelt wurden, nach 20 Jahren bei 24%. Aufgrund von früheren Arbeiten, die jedoch wesentlich kürzere mediane Follow-up-Zeiten als jene von Kiladjian (16,3 Jahre) aufwiesen, wurden wesentlich niedrigere Raten angenommen. Auch wenn mangels Langzeitdaten nicht ausgeschlossen werden kann, dass der natürliche Verlauf einer Polycythaemia vera häufiger als bisher geglaubt zur leukämischen Transformation führt, schloss Petrides mit der Empfehlung, Patienten, die sich schon lange unter einer Therapie mit Hydroxyurea befinden, auf ein Präparat ohne leukämogenes Risiko zu wechseln.
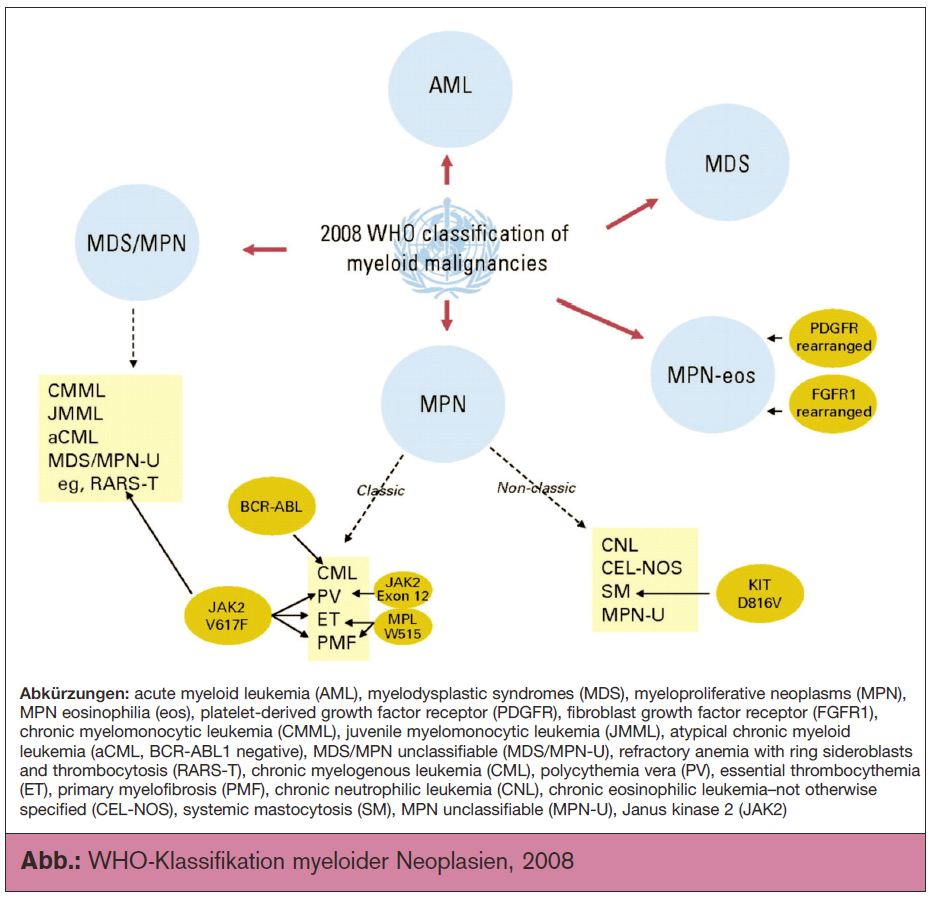
Klinische Relevanz der WHO-Diagnosekriterien
Professor Jürgen Thiele aus Köln wies in seinem Beitrag auf die klinische Relevanz der neuen, im Jahr 2008 herausgegebenen WHO-Diagnosekriterien hin, die nicht zuletzt dank aktualisierter histomorphologischer Kriterien eine bessere Unterscheidung zwischen präfibrotischer primärer Myelofibrose (early PMF) und essenzieller Thrombozythämie (ET) erlauben. In einer rezenten internationalen Studie mit insgesamt 1.071 Patienten (Barbui T. et al., J Clin Oncol 2011; 29:3179) unterschieden sich nach 15 Jahren die beiden Patientengruppen dramatisch bezüglich Gesamtüberleben (ET 80%; early PMF 59%), leukämischer Transformationsrate (ET 2,1%; early PMF 11,7%) und Übergang in eine offenkundige Myelofibrose (ET 9,3%; early PMF 16,9%), wobei die Lebenserwartung der ET-Patienten ähnlich jener der geschlechts- und altersadjustierten europäischen Bevölkerung war. Wie danach Professor Heinz Gisslinger von der Medizinischen Universität Wien betonte, unterscheiden sich diese Entitäten bereits bei Diagnose anhand klinischer Kriterien (Buxhofer-Ausch V. et al., Abstract 510). Zum Beispiel weisen Patienten mit early PMF signifikant höhere Leukozyten-, Plättchenzahlen und LDH-Werte auf als ET-Patienten. Die höheren Leukozytenzahlen dürften bei der early PMF interessanterweise mit einem vermehrten Auftreten von arteriellen Thromboembolien assoziiert sein. Professor Gisslinger wies weiters darauf hin, dass er den Einsatz des, wie erwähnt, potenziell leukämogenen Hydroxyurea speziell bei ET-Patienten mit ihrer weitestgehend normalen Lebenserwartung als Overtreatment betrachtet, zumal in Österreich zur Senkung der Thrombozytenzahlen mit Anagrelid eine Alternative ohne vergleichbare Langzeittoxizität für die Erstlinientherapie zur Verfügung steht. Mit der Wiederentdeckung von Interferon in der Indikation MPN dürfte sich mittelfristig eine weitere therapeutische Therapieoption eröffnen. Aktuell läuft an 6 österreichischen Zentren eine Phase-I-Studie, die die Sicherheit eines neuen pegylierten Interferon α-2b mit besonders langer Halbwertzeit bei Polycythaemia-vera-Patienten untersucht (PEGINVERA, ClinicalTrials.gov identifier: NCT01193699).
Mutationen der Januskinase 2
In seinem Plenarvortrag widmete sich Professor Radek Skoda von der Universität Basel dem Thema MPN und Mutationen der Januskinase 2 (JAK2). Diese Mutationen sind bei einem großen Teil der Patienten mit MPN nachweisbar und führen in letzter Konsequenz über Anstoßen einer Reihe von promitotischen Signaltransduktionswegen zur unregulierten Proliferation der betroffenen hämatopoetischen Stammzellen. Über den so entstehenden Wachstumsvorteil kommt es schlussendlich zur Expansion eines malignen Klons. Auch wenn JAK2-Mutationen nahezu pathognomonisch sind für myeloproliferative Erkrankungen, so besteht Konsens darüber, dass sich die grundlegenden molekulargenetischen Events chronologisch vor der Entstehung der JAK2-Mutationen ereignen. Man spricht heute von sog. “driver mutations”, also Mutationen, die zur Ausprägung eines bestimmten Phänotyps führen (z.B. JAK2) und die von Mutationen abgegrenzt werden, die eher an der Initiation (z.B. TET2, ASLX1) bzw. Progression (z.B. CBL, IKZF1) der molekularen Events Anteil haben dürften. Es scheint weiters eine enge Genotyp-Phänotyp-Beziehung mit unmittelbarer Auswirkung auf die Prognose der individuellen Erkrankung zu bestehen: So ist bei der zumeist benigne verlaufenden essenziellen Thrombozythämie (ET) in der Regel ein viel geringerer Prozentsatz der Granulozyten von MPN-typischen Mutationen betroffen als bei der primären Myelofibrose (PMF) oder der Polycythaemia vera (PV), außerdem betreffen diese Mutationen bei der ET meist nur ein Allel, während speziell JAK2-Mutationen bei PV und fortgeschrittener PMF sehr oft homozygot vorliegen. Neuere Untersuchungsergebnisse deuten weiters darauf hin, dass sich bei den MPN nicht gezwungenermaßen nur jeweils ein maligner Klon mit sequenzieller Akquirierung von Mutationen etabliert. Auch eine Entwicklung zur Biklonalität konnte gezeigt werden. Das heißt, dass verschiedene Stammzellen mit verschiedenen, proliferationssteigernden Mutationen offensichtlich nebeneinander expandieren können. Im Streben nach einem besseren Verständnis für die Chronologie der molekularen Events der MPN dürften laut Professor Radek in nächster Zeit vor allem die relativ seltenen Fälle von familiären MPN ins Zentrum des Interesses rücken.
Internationales Anagrelid-Register
In den anderen MPN-zentrierten Sessions wurde unter anderem betont, dass sich neben den in nächster Zukunft wohl erhältlichen JAK2-Inhibitoren (allen voran Ruxolitinib; die COMFORT-I- und -II-Studienergebnisse wurden unter anderem beim letzten Meeting der European Hematology Association in London präsentiert) vor allem in der Indikation PMF auch andere interessante Substanzen wie zum Beispiel der Immunmodulator Pomalidomid im Stadium der klinischen Erprobung befinden. Ein interessanter heimischer Beitrag war die Präsentation von Daten aus dem internationalen Anagrelid-Register durch Professor Michael Steurer von der Medizinischen Universität Innsbruck. Über 1.500 ET-Patienten aus 10 Ländern wurden bisher in diesem Register mit einem medianen Follow-up von 1,9 Jahren (Range: 10 Tage bis 10 Jahre) verfolgt. Die vorläufigen Ergebnisse zeigen, dass Anagrelid sowohl in der Erst- als auch Zweitlinie nicht nur zu einem guten Ansprechen der Thrombozytenzahlen (Responserate 82 %), sondern auch zu einer dramatischen Abnahme von ET-assoziierten Symptomen führt (0,8% nach vs. 8% vor Therapiebeginn als Erstlinien- bzw. 3% vs. 18% als Zweitlinientherapie), und das bei in Summe sehr gutem Sicherheitsprofil. Ein Übergang in eine Post-ET-Myelofibrose wurde bislang bei weniger als 2% der Patienten gefunden.
Fact BoXx
Das Sicherheitsprofil der in der Behandlung der MPN oft langfristig eingesetzten Substanzen gewinnt immer mehr an Bedeutung. Speziell Hydroxyurea sollte aufgrund seiner potenziellen Leukämogenität bei Patienten unter 60 Jahren nur mehr sehr zurückhaltend eingesetzt werden.
Abhängig von der Art der Zytose kommen Alternativen wie Anagrelid oder pegyliertes Interferon α in Frage, obwohl letzteres für die Therapie der MPN noch keine Zulassung besitzt. Die Mechanismen der MPN-Entstehung werden schrittweise besser verstanden, die grundlegenden molekulargenetischen Events bleiben aber weiterhin unklar.

Ursprünglich erschienen:
UIM 09|2011
UIM 09|2011
Herausgeber: Univ.-Prof. Dr. Günter J. Krejs, Österreichische Gesellschaft für Innere Medizin
Publikationsdatum: 2011-11-11
Zur Ausgabe »
Publikationsdatum: 2011-11-11
Zur Ausgabe »