





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Idiopathische Lungenfibrose – Diagnose und Therapie
24. Mai 2019
Die idiopathische Lungenfibrose (IPF) bezeichnet eine chronisch fibrosierende Lungenerkrankung unklarer Genese, die zumeist im höheren Lebensalter auftritt und zu einem progredienten Lungenvolumsverlust führt. Radiologisch und histologisch ist sie durch das Muster der UIP („usual interstitial pneumonia“) gekennzeichnet. Die IPF gilt mit einer jährlichen Neuerkrankungsrate von 5–20/100.000 als selten. Unbehandelt wurde das mittlere Überleben ab Diagnose mit unter 4 Jahren beziffert, seit einigen Jahren sind aber zwei Präparate verfügbar, die die Progression bremsen und die Mortalität senken können. Leider ist derzeit weiterhin wohl nur ein Bruchteil der erkrankten Personen identifiziert und suffizient therapiert. Es ist daher wichtig, IPF und andere interstitielle Lungenerkrankungen (ILD) als relevante Differenzialdiagnose in der Abklärung von Dyspnoe und Husten zu bedenken.1–3
Symptome und Anamnese
Meist manifestiert sich die IPF schleichend mit zunehmender Belastungsdyspnoe und trockenem Reizhusten.4 Oft werden Patienten aufgrund der unspezifischen Symptome zuvor schon einer kardialen oder HNO-ärztlichen Abklärung unterzogen. IPF-Patienten sind klassischerweise Männer (> 70 %) und zumeist älter als 60 Jahre. Rauchen ist ein Risikofaktor für IPF, was auch zum Teil die Komorbiditäten, wie kardiovaskuläre Erkrankungen und COPD, erklärt.4
Diagnostik der IPF
Auskultation und klinische Untersuchung: Typischerweise kann bei IPF ein basales spätinspiratorisches Knisterrasseln, auch „Sklerosiphonie“ oder „velcro crackles“ („Reißverschlussknistern“) genannt, auskultiert werden. Insbesondere besteht ein Zusammenhang mit dem Vorhandensein von Honigwabenzysten („honeycombing“) in der Computertomografie (CT), die für IPF typisch sind.5
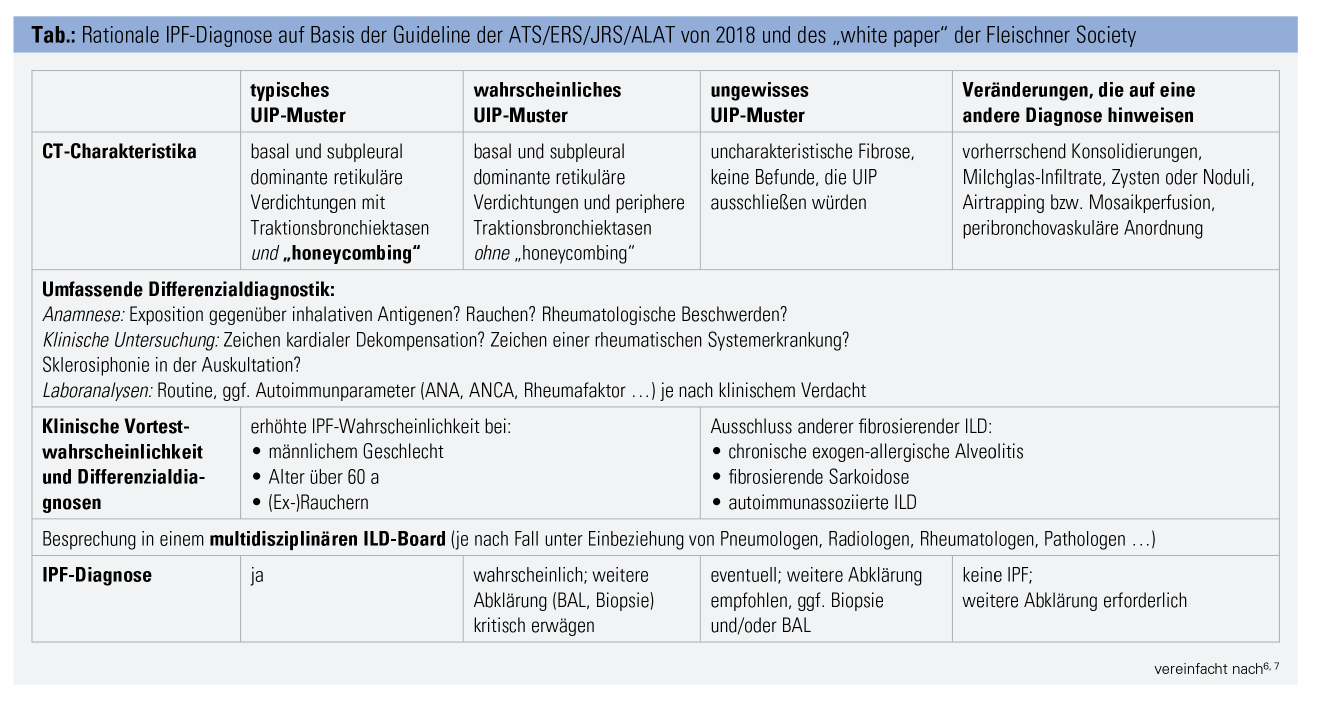
Bei Vorliegen einer Sklerosiphonie und typischer Risikokonstellation (Tab.) sollte an die Diagnose einer IPF gedacht und eine CT-Untersuchung indiziert werden.
In der klinischen Untersuchung sollte zur Differenzialdiagnose auch besonders auf Haut- oder Gelenkveränderungen durch Kollagenosen oder rheumatoide Arthritis geachtet werden, da diese mit ähnlichen interstitiellen Lungenveränderungen einhergehen können.
Computertomografie bei IPF: Radiologischer Goldstandard der IPF-Diagnose ist die HR-(High-Resolution-)CT des Thorax. Diese sollte ohne Kontrastmittel (nativ) und in tiefer Inspiration durchgeführt werden. Wichtig ist, dass die hoch aufgelösten Bilder (Schichtdicken 1 bis 2 mm) nicht nur für die Befundung rekonstruiert, sondern auch dauerhaft gespeichert werden, damit auch für etwaige spätere Fallbesprechungen oder Verlaufskontrollen der gesamte Datensatz vorliegt.6, 7
2018 wurde zunächst ein Positionspapier der Fleischner Society, etwas später auch eine neue gemeinsame Leitlinie der global wichtigsten respiratorischen Gesellschaften veröffentlicht. In beiden wurde die radiologische und histologische Diagnose des UIP-Musters verfeinert. So wird nun zwischen einem definitiven, einem wahrscheinlichen und einem ungewissen („indeterminate“) UIP-Muster unterschieden. Für ein definitives UIP-Muster ist der Nachweis von Honigwabenzysten in basaler und subpleuraler Verteilung (Abb. A) nötig. Können relevante Differenzialdiagnosen wie eine Lungenbeteiligung bei rheumatologischen Erkrankungen ausgeschlossen werden, ist keine histologische Untersuchung zur Diagnose der IPF nötig. Neu ist, dass nun auch bei wahrscheinlichem UIP-Muster in gewissen Fällen die Biopsie unterbleiben kann. Dies ermöglicht auch schon in früheren Stadien eine antifibrotische Therapie einzuleiten, selbst wenn eine Biopsie aus Nutzen-Risiko-Abwägungen heraus nicht möglich erscheint. Empfohlen wird in unklaren Situationen nun auch die Durchführung einer bronchoalveolären Lavage (BAL), da hierdurch mehrere wichtige Differenzialdiagnosen wie exogen allergische Alveolitis oder Sarkoidose risikoarm evaluiert werden können.6, 7

Lungenfunktion bei IPF: Wie bei fibrosierenden interstitiellen Lungenerkrankungen generell, zeigt sich bei IPF gewöhnlich eine pulmonale Restriktion wie auch eine Diffusionsstörung. Speziell die Restriktionsparameter TLC und FVC können aber täuschen, da oft gleichzeitig ein Lungenemphysem vorliegt (gemeinsamer Risikofaktor: Rauchen!), das resultierende Lungenvolumen kann dann „pseudonormal“ sein. Keinesfalls soll jedenfalls bei „zu guter“ Lungenfunktion die Diagnose einer IPF verzögert und eine antifibrotische Therapie vorenthalten werden.8
Laboranalysen bei IPF-Verdacht: Laboranalysen dienen bei IPF einzig der Differenzialdiagnose: Neben den Routineparametern wie Blutbild, Ionogramm, Nieren- und Leberfunktionsparametern empfiehlt sich bei Diagnose ein Screening mit einer begrenzten Auswahl an Autoimmun-Parametern (wie z. B. ANA, ANCA, Rheumafaktor, CCP-Antikörper, ds-DNA-Antikörper). Bei klinischem Verdacht auf spezifische Autoimmunerkrankungen sollte die Testung in enger Absprache mit einem Rheumatologen entsprechend ausgeweitet werden. Wichtig ist, an dieser Stelle anzumerken, dass das UIP-Muster nicht für die IPF spezifisch ist, es kann z. B. auch bei Lungenbeteiligung bei rheumatoider Arthritis vorkommen.6, 7, 9
Therapie der IPF
Nach jahrelangen Irrwegen konnten in den letzten Jahren mit den Substanzen Pirfenidon und Nintedanib erstmals zwei wirksame antifibrotische Therapien etabliert werden. Beide wirken über verschiedene molekulare Wege der überschießenden Vernarbung entgegen. Nintedanib wie auch Pirfenidon sind als Dauertherapie gedacht und müssen täglich eingenommen werden (2-mal 1 bzw. 3-mal 3 Tbl./d). Mögliche Nebenwirkungen beider sind Übelkeit, Inappetenz und Diarrhö, welche durch symptomatische Medikation und kurzfristige Dosisreduktionen zumeist gut kontrollierbar sind. Als spezifische Nebenwirkungen sind vor allem Phototoxizität und Exanthem für Pirfenidon und Leberwerterhöhungen für Nintedanib zu nennen. Beide Substanzen können den jährlichen Verlust an Lungenvolumen um bis zu 50 % hemmen und senken nachweislich die krankheitsspezifische Mortalität, für Nintedanib konnte auch eine Verminderung der zumeist tödlichen akuten Exazerbationen der IPF gezeigt werden.10, 11
Wichtig: Die derzeit verfügbaren Antifibrotika können IPF weder heilen noch zum Stillstand bringen, sondern nur verlangsamen. Essenziell ist daher eine frühe Diagnose und sofortiger Therapiebeginn, denn je mehr Lungenvolumen noch vorhanden ist, desto länger kann die Lebensqualität erhalten werden.
Die Wahl des Präparats sollte in Abwägung von Komorbiditäten, erwartetem Nebenwirkungsprofil und Patientenpräferenz erfolgen. Die Therapieziele und die Prognose sollten offen mit den Patienten diskutiert werden.
Wie auch bei anderen Lungenerkrankungen sollte bei IPF eine Langzeitsauerstofftherapie verordnet werden, wenn indiziert. Ebenso hat die pulmonale Rehabilitation auch bei IPF nachgewiesene positive Effekte und sollte daher jedem Betroffenen angeboten werden. Speziell jüngere Patienten (< 65 Jahre, in Einzelfällen bis ca. 70 Jahre) sollten bei Fehlen von Kontraindikationen früh (!) an einem Lungentransplantationszentrum vorgestellt werden. In fortgeschrittenen Stadien soll auch früh eine palliative, symptomorientierte Therapie etabliert werden.
Resümee
IPF ist eine chronisch progrediente fibrosierende Lungenerkrankung, mit dem Verlust an Lungenvolumen und Diffusionskapazität gehen Leistungsfähigkeit und Lebensqualität verloren. Essenziell ist daher eine frühe Diagnose, die auf der typischen Bildgebung und dem Ausschluss von Differenzialdiagnosen beruht und die interdisziplinär („ILD-board“) gestellt werden sollte. Eine histologische Untersuchung ist hierbei nur in unklaren Fällen notwendig. In Folge sollte unverzüglich eine antifibrotische Therapie begonnen werden. Wichtig ist auch die umfassende Aufklärung der Patienten und ihrer Angehörigen, da die IPF trotz verfügbarer wirksamer Therapien weiterhin eine unheilbare Krankheit mit unabsehbar
em Verlauf darstellt.
1 Lederer DJ et al., N Engl J Med 2018; 378(19):1811–23
2 Gibson GJ et al., Eur Respir J 2013; 42(3):559–63
3 Raghu G et al., Lancet Respir Med 2014; 2(7):566–72
4 Behr J et al., Eur Respir J 2015; 46(1):186–96
5 Sgalla G et al., BMC Pulm Med 2018; 18(1):103
6 Raghu G et al., Am J Respir Crit Care Med 2018; 198(5):e44–e68
7 Lynch DA et al., Lancet Respir Med 2018; 6(2):138–53
8 Kolb M et al., Thorax 2017; 72(4):340–6
9 Fischer A et al., Eur Respir J 2015; 46(4):976–87
10 Richeldi L et al., Respir Med 2016; 113:74–9
11 Noble PW et al., Eur Respir J 2016; 47(1):243–53
_Klinik_fu%cc%88r_Lungenerkrankungen_KUK_Linz_opt.jpg)
Dr. David Lang
Klinik für Lungenheilkunde,Kepler Universitätsklinikum, Linz

Ursprünglich erschienen:
UIM 04|2019 Themenheft Orphan Diseases
UIM 04|2019 Themenheft Orphan Diseases