





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
LAL-Defizienz – Fehldiagnose Fettleber
24. Mai 2019
Der Terminus „Fettleber“ ist ein deskriptiver Begriff, um ein Spektrum unterschiedlicher Lebererkrankungen zu subsumieren. Grundsätzlich kann die Erkrankung als fettige Infiltration im Sinne einer „benignen Steatose“ in Erscheinung treten oder sich aber als nichtalkoholische Steatohepatitis präsentieren, verbunden mit einem deutlich erhöhten Risiko, eine Leberzirrhose oder ein hepatozelluläres Karzinom zu entwickeln. Die Abgrenzung dieser im klinischen Alltag häufigen, alimentär bedingten, auch als „primär“ bezeichnete, Fettlebererkrankung von selteneren „sekundären“ Erscheinungsformen kann durchaus eine Herausforderung darstellen. Hier soll im Speziellen auf eine dieser sekundären, zwar seltenen, aber gut behandelbaren Formen näher eingegangen werden.
Epidemiologie/Pathophysiologie
Die LAL-Defizienz ist eine seltene, autosomal-rezessiv vererbte lysosomale Speichererkrankung mit einer Prävalenz von ca. 1 : 175.000.1 Durch die verminderte Aktivität der lysosomalen sauren Lipase (LAL), welche für die Hydrolyse von Triglyceriden und Cholesterinester in den Lysosomen verantwortlich ist, kommt es zur Akkumulation dieser Substrate insbesondere in Hepatozyten und Makrophagen und in geringerem Maß auch in anderen Zellen des Körpers.2, 3 Die zunehmende lysosomale Lipid-Akkumulation führt zu einer progredienten mikrovesikulären Lebersteatose, welche als inflammatorischer Trigger im Verlauf zur Fibrose und Zirrhose führt.
Klinik
Man differenziert zwischen zwei verschiedenen Erscheinungsformen der LAL-Defizienz, welche sich durch die residuale Enzymaktivität und den klinischen Verlauf unterscheiden.
Die sogenannte Wolman-Krankheit stellt die schwerwiegendere Form dar, beruht auf einem kompletten Fehlen einer LAL-Enzymaktivität und manifestiert sich in den ersten Lebenstagen. Die Krankheit präsentiert sich unter anderem mit einer Hepatosplenomegalie, adrenaler Kalzifikation und gastrointestinaler Symptomatik, welche in einer Gedeihstörung resultiert. Die Erkrankung verläuft katastrophal und führt infolge eines Multiorganversagens innerhalb eines Jahres, meist sogar binnen 3–6 Monaten, zum Tod.4
Die Cholesterinester-Speicherkrankheit (CESD) basiert auf Defekten im gleichen Enzym, das jedoch eine geringe verbleibende Aktivität aufweist. Daher ist die CESD durch einen variablen, milderen Verlauf definiert.5, 6 Die Betroffenen entwickeln sehr unterschiedlich erst im Kleinkind- oder jungen Erwachsenenalter typische Veränderungen. Die klinischen Charakteristika der Erkrankung von 135 Patienten wurden 2013 in einem Review von Bernstein et al.6 zusammengefasst. Typischerweise präsentieren sich die Patienten mit einer Fettleber und einer milden Transaminasenerhöhung (ALT). Weiters zeigt sich meist eine Hepatomegalie, welche bei nahezu allen Patienten, und eine Splenomegalie, welche bei ungefähr 74 % der Betroffenen auftritt.6
Häufig kommt es bereits im jungen Alter zu einer Progredienz der Lebererkrankung über Fibrose und Zirrhose bis zum terminalen Leberversagen. Die Lebererkrankung stellt auch die Haupttodesursache dar, zumeist vor Erreichen des 40. Lebensjahres. Außerdem ist bei so gut wie allen Patienten eine Hypercholesterinämie mit typischerweise erhöhten LDL-Cholesterin- und Triglycerid-Werten bei zugleich erniedrigtem HDL-Cholesterin nachzuweisen, womit das Lipidprofil einer typisch atherogenen Dyslipidämie entspricht, allerdings zumeist bei fehlenden Merkmalen der Insulinresistenz.3 In Fallberichten wurde über atherosklerotische Veränderungen und kardiovaskuläre Events in jungem Alter berichtet, systematische Daten dazu existieren nicht.2 Die klinischen Charakteristika sind in der Tabelle zusammengefasst.

Diagnostik
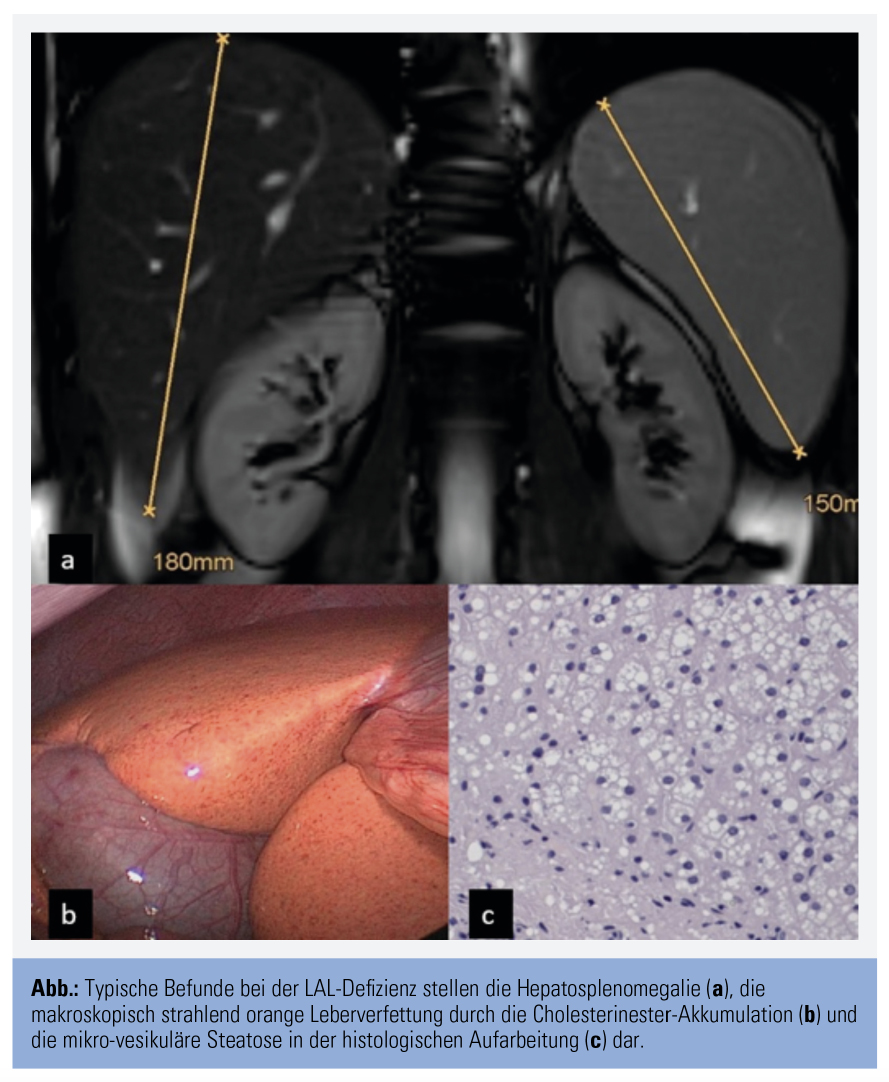
Die LAL-Defizienz stellt eine seltene, aber wichtige Differenzialdiagnose zur nichtalkoholischen Fettlebererkrankung (NAFLD) dar. Insbesondere bei schlanken Patienten ohne klassische Komponenten des metabolischen Syndroms sowie bei jungen Patienten mit fortgeschrittener Lebererkrankung oder Vorliegen einer Hepatosplenomegalie muss an das Vorliegen der Lipidspeichererkrankung gedacht werden.7 Weitere klassische Charakteristika, die auf das Vorliegen der LAL-Defizienz hinweisen können, sind die mikrovesikuläre Leberverfettung in der histologischen Aufarbeitung, welche durch spezielle immunhistochemische Untersuchungen näher charakterisiert werden kann, und die makroskopisch eindrucksvolle orange Farbe des Lebergewebes (Abb.).8, 9 Das Vorliegen des klassischen Lipidprofils der Dyslipoproteinämie Typ 2b mit moderat erhöhten Triglycerid- und LDL-Cholesterin-Werten und zugleich vermindertem HDL-Cholesterin bei Patienten ohne sonstige Komponenten des metabolischen Syndroms stellt ein zusätzliches klinisches Merkmal der Erkrankung dar.
Als Goldstandard in der Diagnostik der LAL-Defizienz gilt aktuell ein Trockenbluttest zur Bestimmung der LAL-Enzymaktivität.10 Dieser erlaubt in einem Referenzlabor (für Österreich AKH Wien, Stoffwechsellabor der Universitätsklinik für Kinder- und Jugendheilkunde) die zuverlässige Unterscheidung von gesunden und erkrankten Individuen. In weiterer Folge kann bei Nachweis der defizienten Enzymaktivität durch Sequenzierung des LIPA-Gens die zugrundeliegende Mutation identifiziert werden. Die häufigste von aktuell 120 verschiedenen bekannten krankheitsverursachenden Mutationen stellt die Deletion in Exon 8 (E8SJM, c.894G>A) dar.1
Therapie
Für lange Zeit stand die Behandlung der Dyslipidämie mittels Statintherapie, Cholestyramin oder Ezetimib im Vordergrund. Dies brachte jedoch nur mäßigen Erfolg bezüglich des Lipidprofils, ohne eine nennenswerte Besserung der Lebererkrankung zu erzielen.6 Weiters wurden einige Patienten beispielsweise nach Auftreten von Lebertumoren oder Leberversagen mittels Lebertransplantation behandelt. Allerdings kam es selbst nach Lebertransplantation zu einem mittelfristig fatalen Verlauf bei systemischer Progredienz der Erkrankung.11 Unlängst wurde mit Sebelipase Alfa eine Enzymersatztherapie zur Behandlung der LAL-Defizienz verfügbar. Hierunter kam es zu einer Besserung der Transaminasen, des Lipidprofils und der Leberverfettung sowie zum Überleben der an der Wolman-Krankheit leidenden Säuglinge über die historische Lebenserwartung hinaus.12, 13
Resümee
Die LAL-Defizienz stellt eine seltene Differenzialdiagnose zur NAFLD dar, die durch einen Enzymdefekt im Lipidstoffwechsel bedingt ist. Typischerweise kommt es zu einer rasch progredienten Lebererkrankung und einem pathologischen Lipidprofil, welches zu einer frühzeitigen Atherosklerose führen kann. Die Diagnose kann mittels Trockenbluttest gestellt werden. Mit Sebelipase Alfa steht eine Enzymersatztherapie zur Behandlung der Erkrankung zur Verfügung.
1 Carter A et al., J Hepatol 2019; 70(1):142–50
2 Beaudet AL et al., J Pediatr 1977; 90(6):910–4
3 Reiner Z et al., Atherosclerosis 2014; 235(1):21–30
4 Jones SA et al., Genet Med 2016; 18(5):452–8
5 Aigner E et al., Am J Gastroenterol 2018; 113(3):443–5
6 Bernstein DL et al., J Hepatol 2013; 58(6):1230–43
7 Strebinger G et al., Lysosomal acid lipase deficiency – early diagnosis is the key. Hepatic Medicine: Evidence and Research. 2019; Accepted Manuscript
8 Hulkova H et al., Histopathology 2012; 60(7):1107–13
9 Zandanell S et al., J Gastrointest Surg 2019; 23(3):601–2
10 Hamilton J et al., Clin Chim Acta 2012; 413(15–16):1207–10
11 Bernstein DL et al., Mol Genet Metab 2018; 124(1):11–9
12 Burton BK et al., N Engl J Med 2015; 373(11):1010–20
13 Jones SA et al., Orphanet J Rare Dis 2017; 12(1):25

AutorIn: Dr. Georg Strebinger
Universitätsklinik für Innere Medizin I, Paracelsus Medizinische Universität Salzburg

AutorIn: Assoz. Prof. Priv.-Doz. Dr. Elmar Aigner
Universitätsklinik für Innere Medizin I, Paracelsus Medizinische Universität Salzburg

AutorIn: Prim. Univ.-Prof. Dr. Christian Datz
Abteilung für Innere Medizin, Krankenhaus Oberndorf, Salzburg

Ursprünglich erschienen:
UIM 04|2019 Themenheft Orphan Diseases
UIM 04|2019 Themenheft Orphan Diseases