





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Lymphangioleiomyomatose und pulmonale Alveolarproteinose
20. März 2020
Als „selten“ werden in Europa Erkrankungen bezeichnet, von denen weniger als 5/10.000 Einwohner betroffen sind. In der Pneumologie gibt es einige der seltenen Erkrankungen, die mehr oder weniger dem Formenkreis der interstitiellen Lungenerkrankungen zugerechnet werden. Seltene Lungenerkrankungen sind jedoch nicht immer nur auf die Lunge beschränkt.
Die wichtigsten Vertreter der seltenen Lungenerkrankungen sind:
- Vaskulitiden (GPA, MPA, EGPA, „Behçet’s disease“, Takayasu)
- Autoimmunerkrankungen (Anti-BM-Syndrom, PAP)
- Erkrankungen mit genetischem Hintergrund (Lymphangioleiomyomatose, Birt-Hogg-Dubé-Syndrom, CF, pulmonale alveoläre Mikrolithiasis)
- primäre ziliäre Dyskinesie
- andere idiopathische Lungenerkrankungen (EOP, Tracheobronchopathia osteochondroplastica, Mounier-Kuhn-Syndrom,idiopathische Bronchiolitis, idiopathische pulmonale Hämosiderose)
- andere seltene Lungenerkrankungen (thorakale Endometriose, PLCH)
Dieser Beitrag beschränkt sich auf zwei seltene Lungenerkrankungen, für die seit einigen Jahren wissenschaftlich fundierte Therapien in speziellen pneumologischen Zentren angeboten werden können: die Lymphangioleiomyomatose und die pulmonale Alveolarproteinose.
Lymphangioleiomyomatose
Die Lymphangioleiomyomatose (LAM) ist eine seltene systemische neoplastische Erkrankung durch Mutationen im TSC1- oder TSC2-Gen; diese Mutationen führen zur mTOR-Aktivierung und zur klonalen neoplastischen Proliferation von „atypischen glatten Muskelzellen“ (LAM-Zellen), welche zu einer Invasion der Lunge führen. Durch die pathologische Proliferation der LAM-Zellen entlang der bronchovaskulären und lymphatischen Strukturen der Lunge kommt es zu einem „remodeling“ und einer zystischen Ausweitung der terminalen Luftwege, Obstruktion und Ausweitung der venösen und lymphatischen Gefäße, Ruptur von Venolen und Lymphgefäßen.
Sporadische vs. mit tuberöser Sklerose assoziierte LAM: Unterschieden wird zwischen einer sporadischen LAM (S-LAM TSC2) und einer mit tuberöser Sklerose assoziierten LAM (TSC-LAM). Die Prävalenz wird auf 3–5/Million geschätzt, bei der S-LAM sind Frauen in der Prämenopause betroffen, selten postmenopausal (hier nimmt man an, dass die Erkrankung sich in milder Form bereits prämenopausal entwickelt hat).
Klinisch präsentieren sich die Patientinnen mit progredienter Belastungsdyspnoe und Reizhusten, bis 40 % geben auch Hämoptysen an. Lungenfunktionell besteht eine obstruktive Ventilationsstörung – auch bei Nichtraucherinnen. Ein Spontanpneumothorax, auch bilateral, findet sich häufig als Erstmanifestation der LAM bei bis zu 40 %, Chylothorax bei bis zu 10 %. Wenn eine LAM suspiziert wird, müssen auch mögliche extrapulmonale Manifestationen bedacht werden: renale Angiomyolipome (bis 60 %), chylöser Aszites, Chyloperikard, Zysten in Leber/Niere/Pankreas, Meningeome, multilokuläre Lymphadenopathien.
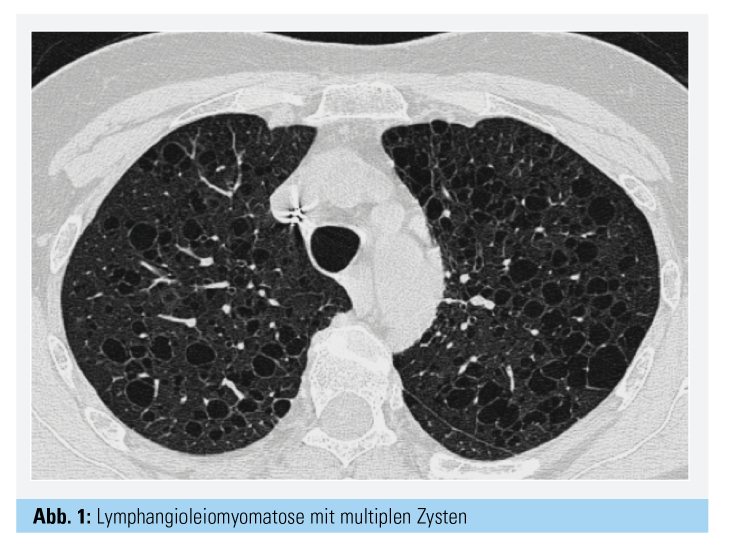
Im Zentrum der Diagnostik steht primär die Bildgebung mit der CT-Thorax (Dünnschicht): Typisch sind dünnwandige, diffus verteilte, rundlich konfigurierte Zysten bis zu 2 cm Durchmesser in beiden Lungen (Abb. 1); in Kombination mit einem Pneumothorax sollte die Diagnose einer LAM dringend suspiziert werden. Eine besondere Bedeutung kommt dem Nachweis von HMB-45-positiven LAM-Zellen in der Histologie zu, wobei in der Regel eine transbronchiale bronchologische Lungenbiopsie ausreichend zum Nachweis derselben ist. Als neues diagnostisches Tool hat sich die Bestimmung des VEGF-D im Serum gezeigt: Die Kombination von VEGF-D > 800 pg/ml und zystischen Läsionen im HR-CT und einer obstruktiven Ventilationsstörung bei einer Frau in der Prämenopause genügt, um die Diagnose einer LAM zu stellen.
Therapeutisch steht heute Sirolimus (Rapamycin-Inhibierung von mTOR) bei LAM zur Verfügung bei Patientinnen mit einem FEV1 > 70 % Soll, sodass für dieses Medikament eine Therapieempfehlung ausgesprochen werden kann.
Pulmonale Alveolarproteinose
Die pulmonale Alveolarproteinose (PAP) ist charakterisiert durch eine Deposition von surfactantähnlichem PAS-positivem lipoproteinösem Material in den Alveolen. Die adulte Form der PAP ist eine Autoimmunerkrankung, assoziiert mit Auto-AK gegen GM-CSF („granulocyte macrophage colony-stimulating factor“), die im Serum und in der bronchioloalveolären Lavage (BAL) nachgewiesen werden können. GM-CSF mediiert die Reifung von funktionierenden Alveolarmakrophagen aus Monozyten; bei Vorliegen von Auto-AK gegen GM-CSF bilden sich unreife/dysfunktionale Alveolarmakrophagen aus, die Surfactant nur unvollständig abbauen können, was wiederum zur Deposition des nicht vollständig abgebauten Surfactant in den Alveolen führt. Bei PAP ist die Lungenarchitektur grundsätzlich nicht gestört, es liegt keine Inflammation vor. Die Prävalenz liegt bei 3–4/Million, häufiger sind Männer betroffen, bis zu 82 % der Erkrankten sind Raucher, das mittlere Alter beträgt 40 Jahre.
Klinisch präsentieren sich die Patienten mit produktivem Husten und progredienter Belastungsdyspnoe, lungenfunktionell besteht meist eine restriktive Ventilationsstörung und vor allem eine Belastungshypoxämie.
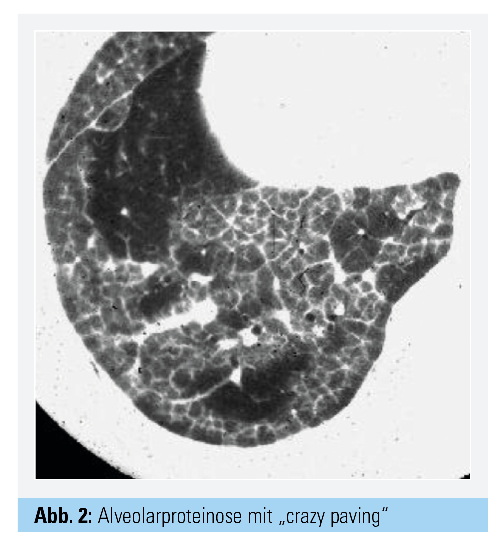
Im Zentrum der Diagnostik steht – wie bei allen ILD – die Bildgebung mittels CT-Thorax (Dünnschicht): Typisch sind geografisch verteilte Milchglastrübungen mit Verdickung der interlobulären Septen, was radiologisch als „crazy paving“ bezeichnet wird (Abb. 2). Wenn die Bildgebung den Verdacht auf eine PAP nahelegt, ist der nächste diagnostische Schritt eine Bronchoskopie mit Durchführung einer BAL: Typischerweise ist die BAL milchig-trüb; wenn die PAS- und Sudanfärbung positiv sind, kann auf eine Lungenbiopsie verzichtet werden, d. h., die Diagnose resultiert aus den Ergebnissen der BAL und der CT-Thorax nach Ausschluss anderer ILD.
Die Therapie der PAP ist abhängig vom Schweregrad der lungenfunktionellen Einschränkung. Selten genügt eine Verlaufskontrolle, in den meisten Fällen ist eine „große Lungenlavage“ die Therapie der Wahl, die nur unter intensivmedizinischen Bedingungen durchgeführt werden kann. Prinzipiell wird bei diesem Verfahren das PAS-positive Material aus den Alveolen „ausgewaschen“. Eine neue vielversprechende Therapieoption ist die Kombination einer Lungenlavage mit der konsekutiven täglichen Inhalation von GM-CSF. Durch diese kombinierte Therapie können wir aktuell Patienten mit dieser seltenen Lungenkrankheit bezüglich ihrer Atemnot und Lungenfunktionseinschränkung besser stabilisieren. Alternativ wird in Fallbeispielen auch die Therapie mit Rituximab und Plasmapherese als Therapieoption beschrieben.
Literatur beim Verfasser

AutorIn: OA Dr. Hubert Koller
Ambulanz für Interstitielle Lungenerkrankungen, 2. Interne Lungenabteilung, Otto-Wagner-Spital, Wien

Ursprünglich erschienen:
UIM 02|2020
UIM 02|2020