






















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Multiples Myelom – Neue Substanzen versprechen Erfolg
23. November 2012
Diagnose: Zur Etablierung der Diagnose eines multiplen Myeloms müssen 2 von 3 der folgenden Kriterien erfüllt sein: Paraprotein-Gradient im Serum und/oder Harn, Vermehrung von monoklonalen Plasmazellen im Knochenmark sowie myelombedingte Knochenveränderungen (Tab. 1). Zu den Standarduntersuchungen zählen: Skelettstatus mittels konventioneller Radiografie, Knochenmarkbiopsie, Serum- und Harn-Elektropherese. Weiters sind weißes und rotes Blutbild, Kalzium, Kreatinin bzw. GFR, Albumin- und Beta-2-Mikroglobulin sowie zytogenetische Untersuchungen (FISH) für die Beurteilung des Krankheitsbildes wichtig.

Mittels Serum- und Harnelektropherese wird das Vorliegen eines Paraproteins bestätigt sowie die Konzentration des Paraproteins bestimmt und mittels Immunfixationselektropherese der Isotyp (IgA, IgG, IgD, IgE, IgM) sowie der Leichtkettentyp (kappa oder lambda) identifiziert. Wichtig für die Prognose- und Verlaufsbeurteilung ist die Bestimmung der freien Leichten sowie deren Verhältnis zueinander (Leichtketten-Ratio) mittels Freelight® Assay.
Die Knochenmarkpunktion aus dem Beckenknochen erlaubt die Gewinnung eines Knochenzylinders sowie die Aspiration von Knochenmark, womit die Infiltrationsdichte mit Myelomzellen und deren morphologischen Charakteristika erfasst werden. Mithilfe der Immunphänotypisierung lässt sich das Verhältnis zwischen klonalen und polyklonalen Plasmazellen bestimmen, was prognostisch relevant ist.
Für den Nachweis von Myelom-bedingten Skelettmanifestationen wird nach wie vor die konventionelle Radiografie des Skelettsystems (Schädel, gesamte WS, Becken, knöcherner Thorax, Humeri und Femura) als Goldstandard herangezogen. Bei extramedullären Myelommanifestationen, drohendem Querschnitt, und schmerzhaften Skelettregionen, die durch konventionelle Röntgenuntersuchungen nicht definitiv abgeklärt werden können, sollte eine MRT-Untersuchung erfolgen. Insgesamt verfügen MRT und CT über eine höhere Sensitivität zum Nachweis von Skelettläsionen. Mittels Positronen-Emissions-Tomografie (PET) kann die Aktivität von Myelom-Manifestationen bestimmt werden, was sowohl zum Therapiebeginn als auch nach etwa 4 Zyklen Chemotherapie relevant ist, da die Erreichung eines negativen PET-Befundes mit einer günstigen Prognose assoziiert ist.
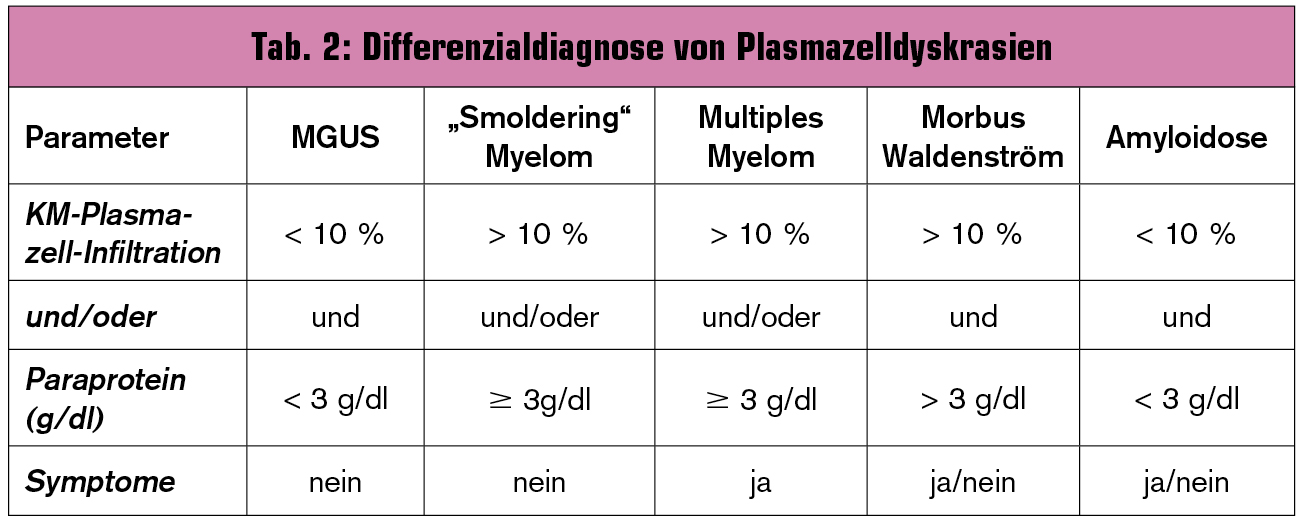
Die Abgrenzung eines multiplen Myeloms von anderen Plasmazellerkrankungen kann manchmal schwierig sein, weswegen die differenzialdiagnostischen Kriterien in Tab. 2 detailliert angeführt sind.
Stadieneinteilung und Risikostratifizierung: Die Stadieneinteilung erfolgt gemäß dem International Staging System (ISS). Dabei werden an Hand von β2-Mikroglobulin und Albumin drei Stadien mit deutlich unterschiedlicher Prognose unterschieden. Durch Einbeziehung weiterer Parameter, wie der Ratio der freien Leichtketten, zytogenetischer Parameter (FISH) oder Genexpressionsmuster kann die Risikostratifizierung weiter verbessert werden. Zytogenetische Untersuchungen (FISH) sollten zum Beginn der Behandlung, und in bestimmten Fällen auch bei neuerlicher Progression der Erkrankung durchgeführt werden, da im Rahmen des Krankheitsverlaufs weitere genetische und zytogenetische Veränderungen auftreten. Durch den Einsatz von CT, MRT sowie PET konnte die Sensitivität zum Nachweis von myelombedingten Skelett- und extraskelettalen Veränderung erhöht werden. Damit kann die Ausdehnung des Krankheitsprozesses besser beurteilt werden.
Therapieindikation: Die Indikation zur Therapie stellt sich bei Vorliegen von Myelom-bedingten Symptomen bzw. bei Erfüllung eines oder mehrerer der in Tab. 3 angeführten CRAB-Kriterien. Derzeit wird in Studien geprüft, ob bei Patienten mit „smouldering“ (schwelendem) Myelom und einem hohen Risiko für einen Übergang in ein aktives Myelom eine präemptive Behandlung (noch vor Auftreten von Symptomen) Vorteile erbringt. Die Ergebnisse einer noch nicht publizierten Studie scheinen die Sinnhaftigkeit dieser Strategie zu bestätigen.

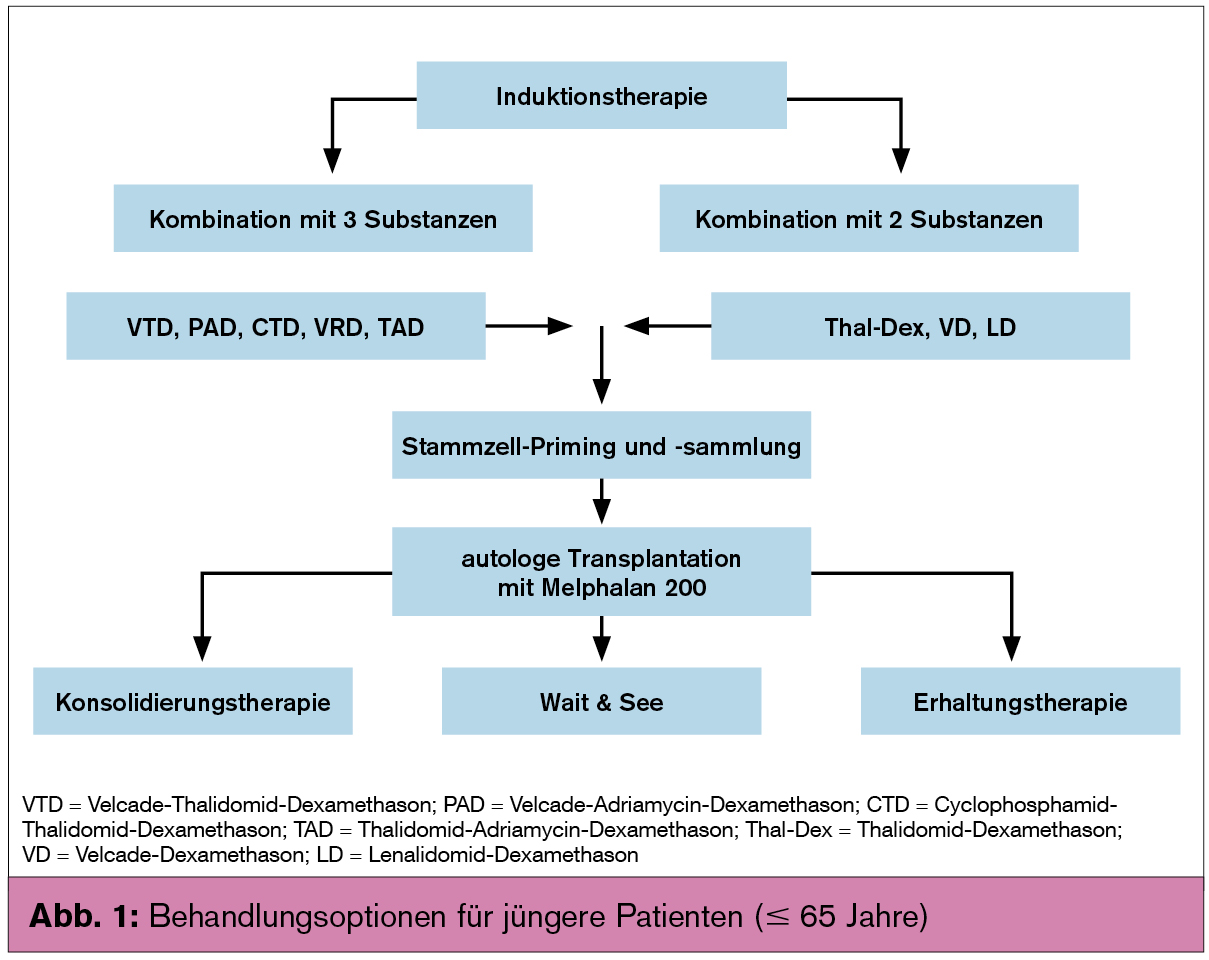
Therapie: Bei jüngeren Patienten (≤ 65 Jahre) sollte nach einer kurzen Induktionstherapie eine autologe Stammzelltransplantation durchgeführt werden, während bei allen anderen Patienten eine konventionelle Chemotherapie eingeleitet werden soll. Zur Induktionstherapie sollten 3–4 Zyklen eines aus 3 Substanzen bestehenden Therapieregimes zur Anwendung kommen; die Zugabe einer 4. Substanz führt zu keiner Verbesserung der Therapieergebnisse, aber zu erhöhter Toxizität. Anschließend erfolgt Gewinnung von Stammzellen und danach die Hochdosistherapie mit Melphalan 200 mg/m2, gefolgt von der Rücktransfusion der autologen Stammzellen (Abb. 1). Mit dieser Therapie werden ein medianes progressionsfreies Überleben von 20–30 Monaten und ein medianes Gesamtüberleben von 6–8 Jahren erreicht.
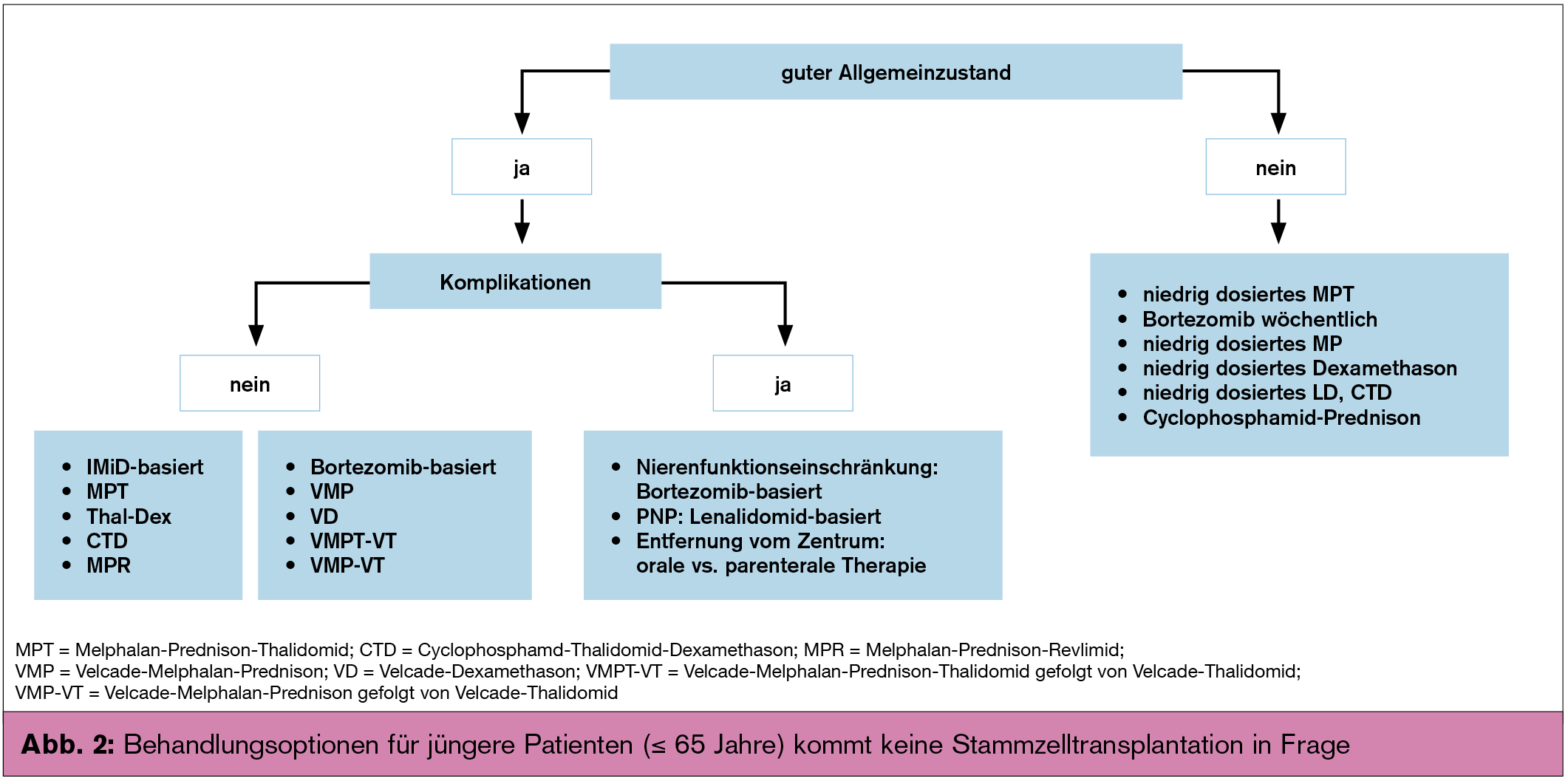
Kommt eine Stammzelltransplantation nicht in Frage, empfiehlt sich die Einleitung einer Chemotherapie, die zumindest eine der neuen Substanzen (Thalidomid, Bortezomib und Lenalidomid) beinhalten sollte (Abb. 2). Bei Vorliegen eines hohen zytogenetischen Risikoprofils, wie Deletionen an Chromosom 17 (del 17p) oder Chromosom 1 (del 1p21) oder Translokation t(4;14) und Amplifikation 1q, sollte Bortezomib-basierten Kombinationen der Vorzug eingeräumt werden. Lenalidomid sollte aufgrund seiner fehlenden Neurotoxizität bei präexistenter Neuropathie zum Einsatz kommen. Wissenschaftlich offen bleibt die Frage nach der optimalen Anzahl von Behandlungszyklen in der Erstlinientherapie. Von den meisten Gruppen wird die Verabreichung von 9–12 Zyklen unabhängig von der Qualität des Ansprechens empfohlen. Bei älteren und insbesondere geriatrischen Patienten müssen Therapie und Dosis an den Gesamtzustand des Patienten angepasst werden.
Konsolidierung und Erhaltungstherapie: Unter Konsolidierungstherapie wird eine intensive, auf 2–4 Zyklen begrenzte Behandlung mit dem Ziel, die Remissionsqualität zu verbessern, verstanden. Bisherige Studienergebnisse zeigen eine deutliche Erhöhung der Zahl der Patienten mit molekularer Remission, was mit einem besonders langen Überleben vergesellschaftet ist. So konnte in einer Studie mit 4 Zyklen einer Velcade®-Dexamethason-Thalidomid-(VDT)-Konsolidierungstherapie (nach autologer Transplantation) die Rate an molekularen Remissionen von 3 auf 18 % erhöht werden.
Mit einer Erhaltungstherapie wird eine Verlängerung des durch die Initialbehandlung erzielten Behandlungserfolgs angestrebt, wobei natürlich nicht nur eine Verlängerung des progressionsfreien, sondern auch des Gesamtüberlebens angestrebt wird. Prinzipiell stehen dafür Thalidomid, Bortezomib und Lenalidomid zur Verfügung. Für Bortezomib liegen keine direkten Vergleiche vor. Mit einer Thalidomid-Erhaltungstherapie kann sowohl das progressionsfreie als auch das Gesamtüberleben verlängert werden. Allerdings wird die Thalidomid-Erhaltungsbehandlung wegen der häufig schlechten Toleranz und der nur in einigen Studien beobachteten Verlängerung des Gesamtüberlebens kaum eingesetzt. Lenalidomid konnte in 3 Studien eine Verdoppelung des progressionsfreien Überlebens und in einer Untersuchung eine Verlängerung des Gesamtüberlebens zeigen. Aufgrund einer geringen Erhöhung der Inzidenz von Zweitmalignomen und der uneinheitlichen Ergebnisse bezüglich Gesamtüberlebens kann derzeit keine generelle Empfehlung für eine Lenalidomid-Erhaltungsbehandlung abgegeben werden.
Rezidivbehandlung: Bei der Wahl der bestgeeigneten Rezidivbehandlung sind patienten-, tumor- und therapiespezifische Faktoren zu berücksichtigen. Patientenalter, Polymorbidität, insbesondere präexistente Polyneuropathie, Performance-Status, Nähe zum Behandlungszentrum sind relevante patientenspezifische Faktoren. Die Biologie der Erkrankung lässt sich aufgrund zytogenetischer Befunde, des progressionsfreien Intervalls, der LDH und Tumorlast erfassen, während die Qualität des Ansprechens auf die Vortherapie sowie die Toleranz und die bereits eingesetzten bzw. noch verfügbaren nicht zuvor verwendeten Substanzen wichtige Aufschlüsse über die Therapieoptionen erlauben. Prinzipiell kann nach längerer progressionsfreier Überlebenszeit eine erfolgreiche Vortherapie wieder eingesetzt werden, während bei ungenügendem Erfolg eine Therapie mit Medikamenten einer anderen Substanzgruppe herangezogen werden sollte.
Neue Therapieentwicklungen: Carfilzomib, ein wirksamer Proteasom-Inhibitor, der keine oder nur geringe Neurotoxizität aufweist und zu hohen Ansprechraten führt, ist bereits in den USA, jedoch noch nicht in Europa zugelassen. Pomalidomid zählt zur Substanzklasse der IMiDs (IMiDs = immuno-modulatory drugs, Immunmodulatoren), ist sehr wirksam und nicht neurotoxisch. Die Zulassung in Europa wird aber noch auf sich warten lassen. Elotuzumab, ein monoklonaler Antikörper gegen CS1 (auf den meisten Myelomzellen exprimiert), könnte das „Rituximab der Myelomtherapie“ werden, da die laufenden Studien eine Wirkungsverstärkung der Chemotherapie zeigen. Weitere erfolgversprechende neue Substanzen sind unter anderem Tyrosinkinase-Inhibitoren (Masitinib), andere monoklonale Antikörper (Eclotuzumab) und Inhibitoren von Adhäsionsmolekülen, die zur Abnabelung von Myelomzellen von der für ihr Überleben wichtigen Bindung ans Stroma und damit zur spontanen Apoptose oder zumindest verstärkten Chemosensitivität der Myelomzellen führen.

AutorIn: Prim. Univ.-Prof. Dr. Heinz Ludwig
1. Medizinische Abteilung – Zentrum für Onkologie und Hämatologie , Wilhelminenspital der Stadt Wien

Ursprünglich erschienen:
UIM 08|2012
UIM 08|2012