






















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Leber und Lunge
11. November 2011
Sowohl HPS und PoPH bleiben derzeit häufig undiagnostiziert; meist werden diese Erkrankungen erst im Zuge einer Lebertransplantations- Evaluierung entdeckt, wo sie bei schwerer Ausprägung durchaus zum limitierenden Faktor werden können. Eine Frühdiagnose erscheint daher wünschenswert, zumal wirksame Therapien vorhanden sind.
Hepatopulmonales Syndrom
Das hepatopulmonale Syndrom (HPS) ist definiert durch die Trias Lebererkrankung, arterielle Deoxyge nierung und intrapulmonale Gefäßdilatation (Tab. 1). Meist liegt eine chronische Lebererkrankung (Leberzirrhose) zugrunde. Für die Hypoxämie existieren unterschiedliche Kriterien (“liberales” Kriterium AaDO2, “strenges” Kriterium paO2). Die pulmonalvaskuläre Dilatation kann entweder durch ein pathologisches Kontrastmittelechokardiogramm oder eine pathologische extrapulmonale Shuntfraktion in der Lungenperfusionsszintigraphie dokumentiert werden.
Der Schweregrad eines HPS wird anhand einer Empfehlung der European Respiratory Society (ERS) Task Force je nach Grad der Hypoxämie wie folgt unterteilt:
-
geringgradiges HPS (paO2 > 80 mmHg)
-
mittelgradiges HPS (paO2 60-80 mmHg)
-
schweres HPS (paO2 < 60 mmHg)
-
sehr schweres HPS (paO2 < 50 mmHg)
Die Prävalenz des HPS variiert zwischen 15 und 30% abhängig von der untersuchten Population, aber auch von der verwendeten Definition der Hypoxämie (30% bei AaDO2 > 15, aber nur 15% bei der strengeren Definition paO2 < 70 mmHg). Schenk et al. fanden in einem österreichischen Kollektiv von 111 Lebertransplantations-Kandidaten am AKH Wien eine Prävalenz von 24%, wobei das Vorhandensein eines HPS die Prognose deutlich verschlechterte (medianes Überleben mit HPS 11 Monate, ohne HPS 41 Monate). Der Verlauf ist üblicherweise progredient mit zunehmender Hypoxämie.
Pathogenetisch liegt eine Dilatation der Pulmonalgefäße zugrunde, welche infolge längerer Diffusionswege den Gasaustausch behindert. Die Mediatoren dieser Vasodilatation sind unbekannt, diskutiert wird eine hepatische Überproduktion von Endothelin 1 (ET-1) bzw. eine Aufregulierung von vasodilatorischen ETB-Rezeptoren.
Zu den wichtigsten klinischen Symptomen zählen (Belastungs-)Dyspnoe, Platypnoe/Orthodeoxie (Dyspnoe bzw. Abnahme der Sauerstoffsättigung beim Lagewechsel vom Liegen zum Sitzen), Trommelschlägelfinger und Uhrglasnägel.
Die Diagnose basiert auf dem Nachweis der Deoxygenierung mittels arterieller Blutgasanalyse (paO2 < 70 mmHg oder Erhöhung der altersentsprechenden AaDO2) und dem Nachweis der intrapulmonalen Gefäßdilatation, entweder mittels Kontrastechokardiographie (intravenöse Injektion von agitierter Saline oder Gelofusin, Kontrast im Linksherz nach > 3 Herzschlägen) oder mittels Lungenperfusionsszintigraphie und Berechnung der extrapulmonalen Shuntfraktion anhand des Hirn- Lungen-Quotienten (normal < 6%) nach der Formel:
| GMT brain / 0,13 GMT brain / 0,13 + GMT lung |
Als einfache und nichtinvasive Screening-Methode ist die Pulsoximetrie zu empfehlen, bei SpO2 < 97% wird die weitere Abklärung wie oben angeführt empfohlen.
Therapie: Die Lebertransplantation (LTX) ist die einzige gesicherte Therapie und führt idealerweise zur Normalisierung der pulmonalen Gasaustauschstörung. Das Vorliegen eines HPS per se kann bereits eine LTX-Indikation darstellen. Da die Prognose der zugrunde liegenden Leberzirrhose durch ein gleichzeitig bestehendes HPS verschlechtert wird (siehe oben), ist eine höhere Dringlichkeitsstufe auf der Leberwarteliste zu fordern. Allerdings steigt das Risiko der LTX beträchtlich bei schwerem HPS mit paO2 20%.
Eine gesicherte Pharmakotherapie des HPS existiert bis dato nicht. Bei schwerer Hypoxämie (paO2 < 60 mmHg) ist eine Heimsauerstofftherapie prinzipiell indiziert (Long- Term Oxygen Therapy – LTOT). Der Einfluss einer LTOT auf die zugrunde liegende Lebererkrankung und die Prognose ist derzeit unbekannt und Gegenstand einer laufenden europäischen Multicenterstudie (LIVAIR Study).
| Tab. 1: Definition des hepatopulmonalen Syndroms (HPS) |
|---|
|
Portopulmonale Hypertonie (PoPH)
Die portopulmonale Hypertonie (PoPH) ist definiert als pulmonalarterielle Hypertonie (PAH) mit erhöhtem Lungengefäßwiderstand (PVR) bei Patienten mit portaler Hypertension ohne andere Ursachen für eine PAH (Tab. 2).
Der Schweregrad einer PoPH wird anhand der ERS Task Force je nach mittlerem pulmonalarteriellen Druck (mPAP) wie folgt unterteilt:
-
milde PoPH (mPAP < 35 mmHg)
-
moderate PoPH (mPAP 35-50 mmHg)
-
schwere PoPH (mPAP > 50 mmHg)
Die Prävalenz der PoPH variiert zwischen 2% und 16% aller Patienten mit portaler Hypertension. In zwei großen Kollektiven an LTXReferenzzentren (USA, Frankreich) fand sich eine PoPH in 5-6%, dies gehäuft bei Patienten mit Autoimmunhepatitis als Ätiologie der zugrunde liegenden Lebererkrankung. Interessanterweise korreliert der Schweregrad der Lebererkrankung (anhand von Child-Pugh-Score oder MELD) nicht mit dem Vorliegen einer PoPH.
Die wichtigsten Faktoren für die Entstehung einer PoPH sind hyperdyname Zirkulation (wie sie bei portaler Hypertension üblicherweise vorliegt), zirkulierende vasoaktive Substanzen wie ET-1 oder asymmetrisches Dimethylarginin (ADMA; ein Inhibitor der endothelialen NO-Synthase) sowie genetische Faktoren (BMPR2, ALK-1). Histopathologisch findet sich ein Remodelling der Pulmonalgefäße wie bei idiopathischer PAH.
Die führenden Symptome sind Belastungsdyspnoe im Frühstadium sowie Thoraxschmerz, Synkopen und periphere Ödeme in fortgeschrittenen Stadien. Die Prognose einer unbehandelten PoPH ist schlecht (medianes Überleben 6 Monate) und hängt sowohl von der kardiopulmonalen Situation als auch vom Grad der Leberdysfunktion ab. Wichtig ist die Suche nach einer allfälligen PoPH im Zuge einer LTX-Vorbereitung, da bei schwerer PoPH (mPAP > 50 mmHg) die perioperative Mortalität massiv erhöht ist. Nach LTX kann sich die PoPH bessern, stabilisieren oder auch verschlechtern. Anhand mehrerer Fallberichte wurde auch das Auftreten einer De-novo-PoPH nach LTX dokumentiert.
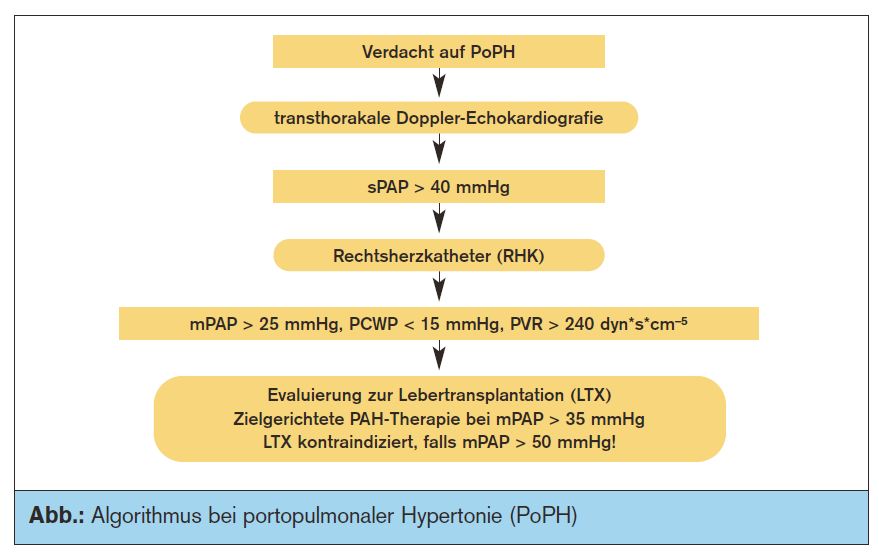
Diagnose: Die wichtigste Screeninguntersuchung ist die transthorakale Doppler-Echokardiographie mit Abschätzung des systolischen pulmonalarteriellen Drucks (sPAP) anhand der Geschwindigkeit des Regurgitations-Jets bei Trikuspidalinsuffizienz. Dabei kommt die vereinfachte Bernoulli-Gleichung zur Anwendung (sPAP = 4v2 + RAP). Falls der so geschätzte sPAP einen Wert von 40-50 mmHg übersteigt, ist zur Bestätigung der Diagnose ein Rechtsherzkatheter mit Messung der hämodynamischen Parameter (mPAP, PCWP, RAP, CO, PVR) indiziert. Beim Cut-off > 50 mmHg hat der nicht-invasiv abgeschätzte sPAP eine hohe Vorhersagekraft (Sensitivität 97%, Spezifität 77%) für eine zumindest mittelgradige PoPH.
Um die Belastbarkeit von Patienten mit PoPH zu dokumentieren, sollten eine Spiroergometrie und ein 6-min-Gehtest durchgeführt werden.
Therapie der PoPH: Für eine zielgerichtete Therapie der pulmonalen Hypertension kommen (inhalative) Prostaglandine, ET-Rezeptor-Antagonisten (ERA) und Sildenafil in Frage. Dabei muss betont werden, dass die Evidenz für den Einsatz bei PoPH nur auf Fallberichten bzw. Fallserien beruht; in den randomisierten Zulassungsstudien dieser Substanzen waren Patienten mit Leberdysfunktion nämlich ausgeschlossen. Allerdings sind es gerade Patienten mit fortgeschrittener Lebererkrankung, welche von einer Anti-PAH-Therapie besonders profitieren können: LTX-Kandidaten mit moderater oder schwerer PoPH, bei denen das perioperative Risiko erst nach erfolgreicher Behandlung der PAH auf ein akzeptables Maß gesenkt werden kann (Abb.).
Bosentan (ein nicht-selektiver ERA mit potenzieller Hepatotoxizität) erzielte in einer Fallserie von 11 Patienten mit Child-A-Zirrhose eine Reduktion der PVR und Besserung der Symptome und Belastbarkeit, dies ohne Verschlechterung der Leberfunktion. Rezente Daten zu Ambrisentan an 13 Patienten (11 Zirrhotiker, davon 8 mit Child A) zeigten ebenfalls anhand hämodynamischer Parameter eine gute Wirksamkeit bei fehlender Hepatotoxizität. Auch intravenöses Epoprostenol bzw. inhalatives Iloprost, Sildenafil sowie diverse Kombinationen der genannten Substanzen konnten die pulmonale Hämodynamik und Belastbarkeit verbessern und in einzelnen Fällen schwerer PoPH eine erfolgreiche Überbrückung zur LTX ermöglichen.
Im Falle einer pfortaderdrucksenkenden Therapie muss der Effekt auf die PAH berücksichtigt werden. So wurde für Terlipressin ein günstiger Effekt auf den PAP beschrieben, während Propranolol die pulmonale Hämodynamik und Belastbarkeit offenbar verschlechtert. Eine TIPS-Implantation gilt bei PoPH aufgrund der damit verbundenen Zunahme der hyperdynamen Zirkulation als relativ kontraindiziert.
| Tab. 2: Definition der portopulmonalen Hypertonie (PoPH) |
|---|
|
|
Fact-Box
|
Literatur beim Verfasser

Ursprünglich erschienen:
UIM 09|2011
UIM 09|2011
Herausgeber: Univ.-Prof. Dr. Günter J. Krejs, Österreichische Gesellschaft für Innere Medizin
Publikationsdatum: 2011-11-11
Zur Ausgabe »
Publikationsdatum: 2011-11-11
Zur Ausgabe »