





















































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Zielgerichtete Behandlung der CLL
28. Mai 2021
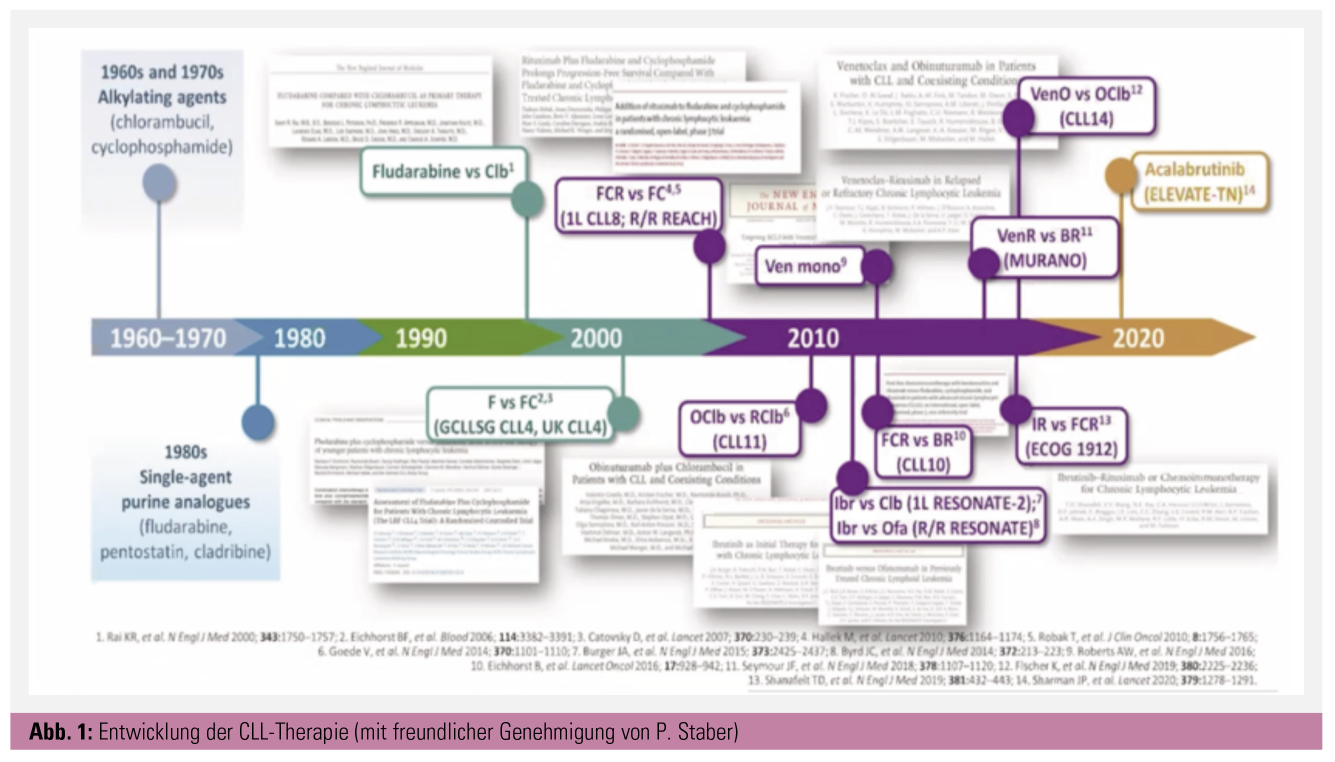
Die chronische lymphatische Leukämie (CLL) ist die häufigste Leukämie des erwachsenen Menschen (Inzidenz: 4–6/100.000). Bis vor etwa 15 Jahren bestand die Behandlung aus ungerichteter Mono- oder Polychemotherapie. Danach stand für nahezu 10 Jahre die Chemoimmuntherapie im Fokus. Mittlerweile stehen zielgerichtete Therapien im Zentrum der risikostratifizierten Behandlungsstrategien (Abb. 1).
Grundlagen
Schon vor längerer Zeit wurden bei der CLL genetische Risikofaktoren detektiert, die mit der damals üblichen Chemotherapie nicht zufriedenstellend behandelbar waren. Es konnte gezeigt werden, dass Alterationen des am kurzen Arm des Chromosom 17 gelegenen p53-Tumorsuppressorgens (Deletionen und/oder Mutationen) mit einer schlechten Prognose assoziiert sind (Doehner et al.). Bei der Erstlinienbehandlung beträgt der Prozentsatz dieser Patienten ungefähr 8–10 %, im Rezidiv entsprechend mehr. Ebenso ist das Vorliegen eines komplexen Karyotyps für ein verkürztes Überleben verantwortlich. Hier ist die Häufigkeit in der Primärtherapie ca. 15 %. Zudem hatten Patienten mit unzureichenden somatischen Hypermutationen im Immunglobulin-Schwerketten-Locus (IgHV unmutiert, Hamblin et al.), die bei therapienaiven behandlungspflichtigen Patienten etwa 60 % ausmachen, unter Chemotherapie ein verkürztes progressionsfreies Überleben und auch Gesamtüberleben.
Chemoimmuntherapie
Mit der Einführung der zu diesem Zeitpunkt revolutionären Chemoimmuntherapie (CIT) FCR (Fludarabin, Cyclophosphamid und Rituximab) durch die deutsche CLL-Studiengruppe (CLL8-Studie, Hallek et al.) und das MD Anderson Cancer Center konnte die Prognose dieser Patienten zwar verbessert werden, dennoch schnitten sie nach wie vor signifikant schlechter ab. Patienten ohne die oben erwähnten Risikofaktoren hatten mit dieser 6-monatigen Behandlung allerdings eine sehr gute Prognose, v. a. wenn sie nach Therapieende keine minimale Resterkrankung mehr aufwiesen (MRD-negativ). Weiterentwicklungen des doch ziemlich nebenwirkungsreichen FCR-Protokolls, im Speziellen die Anpassung des Chemotherapie-Backbones an das Alter und die Komorbiditäten der Patienten mittels der CLL10-Studie (Bendamustin, Eichhorst et al.) oder die Intensivierung des Anti-CD20-Antikörpers bei gleichzeitiger Mitigation der Chemotherapie mittels Chlorambucil in der CLL11-Studie (Obinutuzumab, Goede et al.), konnten die ungünstige Prognose dieser genetischen Veränderungen nicht umkehren.
Zielgerichtete Therapie
Daher bedurfte es der Entwicklung neuer therapeutischer Konzepte für diese Hochrisikogruppe, wobei sich u. a. die Bruton’sche Tyrosinkinase (BTK), die Phosphatidylinositol-3-Kinase (PI3) und das B-Cell-Lymphoma-2-Protein (BCL-2) als attraktive Targets erwiesen. Während die beiden Kinasen bei der CLL eine wesentliche Rolle in der Signaltransduktion des konstitutiv aktivierten B-Zell-Rezeptors spielen, ist BCL-2 ein in den Mitochondrien hochregulierter, essentieller antiapoptotischer Faktor. Phase-1- und -2-Studien mit entsprechenden Inhibitoren, Ibrutinib (BTK, Byrd et al.), Idelalisib (PI3, Sharman et al.) und Venetoclax (BCL2, Roberts et al.), die bei größtenteils rezidivierten Patienten mit erhöhtem genetischen Risiko durchgeführt wurden, zeigten ermutigende Ergebnisse. Die Patienten hatten ein wesentlich längeres progressionsfreies Überleben (PFS) als mit den damaligen Standardtherapien. Daneben war der IgHV-Mutationsstatus für die Prognose nicht länger relevant und auch 17p-alterierte Patienten hatten ein deutlich besseres Outcome als zuvor. Interessanterweise wurden neue substanzspezifische Nebenwirkungen beobachtet. Unter Ibrutinib traten vermehrt Vorhofflimmern, arterielle Hypertonie, Infektionen und milde Blutungen auf. Idelalisib führte häufiger zu Durchfällen, opportunistischen Infektionen und protrahierten immungetriggerten Entzündungen (Kolitis, Pneumonitis und Transaminitis). Bei Venetoclax standen eine initiale Tumorlyse, die das mittlerweile gebräuchliche langsame Eindosieren zur Folge hatte, und die Neutropenie im Fokus.
Als nächster Schritt erfolgten randomisierte Phase-3-Studien bei vorbehandelten Patienten, die diese Ergebnisse im Wesentlichen bestätigten und zur Zulassung dieser Substanzen in der Monotherapie oder in Kombination mit Rituximab führten. Ibrutinib war Ofatumumab überlegen (Resonate-Studie, Byrd et al.), Idelalisib mit Rituximab effektiver als Rituximab in der Monotherapie (Furman et al.) und Venetoclax mit Rituximab signifikant besser als Rituximab/Bendamustin (Murano, Seymour et al.). Erwähnenswert ist, dass im Rezidiv neben dem PFS auch das Gesamtüberleben signifikant verbessert werden konnte. Während die Kinaseinhibitoren nahezu nie zu MRD-Negativität führten und demzufolge als Dauertherapie verabreicht wurden, konnte Venetoclax nach willkürlich gewählten 2 Jahren beendet werden. Hier lagen speziell im Murano-Trial die MRD-Raten bei beeindruckenden 60 %.
Die weitere Entwicklung dieser Substanzen bei nicht vorbehandelten Patienten ließ nicht lange auf sich warten. Hier mussten bezüglich dem PI3K-Inhibitor Idelalisib deutlich erhöhte schwere Toxizitätsraten konstatiert werden, die zum Stopp des weiteren Entwicklungsprogrammes führten. Idelalisib ist heute nur noch als Reservemedikament nach Versagen von BTK-Inhibitoren und Venetoclax zu sehen.
Das Erstlinienstudienprogramm von Ibrutinib sah direkte Vergleiche mit Chemoimmuntherapie für alle möglichen Patiententypen vor: für ältere und/oder komorbide Patienten wurde Chlorambucil (Resonate-2, Burger et al.) oder Obinutuzumab/Chlorambucil (iLLUMINATE, Moreno et al.) als Referenzarm gewählt, wobei in der iLLUMINATE-Studie Ibrutinib mit Obinutuzumab kombiniert wurde. Für die älteren, jedoch sonst fitten Patienten wurde der BTK-Inhibitor mit Rituximab/Bendamustin verglichen (Alliance 202, Woyach et al.), bei den jungen fitten Patienten wurde FCR von Ibrutinib/Rituximab gechallenged (ECOG 1912, Shanafelt et al.). Alle diese Studien zeigten einen höchst signifikanten PFS-Vorteil für die dauerhafte BTK-Inhibition, wobei die Zugabe von Rituximab keinen zusätzlichen Vorteil aufwies. In den Studien, die 17p-Patienten eingeschlossen hatten, war Ibrutinib deutlichst effektiver als die Chemoimmuntherapie, ebenso wie bei den IgHV-unmutierten Patienten. Andererseits liefen die IgHV-mutierten Patienten mit Chemoimmuntherapie genauso gut wie mit dem BTK-Inhibitor. Interessanterweise gab es nur in der ECOG-1912-Studie einen auf wenigen Ereignissen basierenden Gesamtüberlebensvorteil. Zudem konnte man bei der Kombination Obinutuzumab/Ibrutinib in der iLLUMINATE-Studie eine beachtenswerte MRD-Rate von etwa 35 % beobachten. Schließlich stoppten deutlich mehr Patienten den BTK-Inhibitor aufgrund von Nebenwirkungen als wegen Progression.
Oft zeigten sich spezifische Mutationen für die Resistenz verantwortlich. Ibrutinib ist nunmehr als Monotherapie oder in Kombination mit Obinutuzumab für die Erstlinientherapie der CLL zugelassen. Mittlerweile ist auch der Zweitgenerations-BTK-Inhibitor Acalabrutinib für alle CLL-Patienten zugelassen. Acalabrutinib ist ähnlich effektiv wie Ibrutinib, jedoch aufgrund geringerer Off-Target-Effekte besser verträglich – besonders im Hinblick auf die kardiovaskulären Toxizitäten. Dafür können bei dieser Substanz bei ca. 30–40 % der Patienten Kopfschmerzen als neue Nebenwirkung auftreten, die jedoch gut mit koffeinhaltigen Getränken oder Medikamenten behandelbar sind.
Die zulassungsrelevante Venetoclax-Studie ist die CLL14-Studie der deutschen CLL-Studiengruppe, die speziell komorbide Patienten berücksichtigte und Venetoclax-Obinutuzumab mit dem Standard aus der CLL11-Studie Chlorambucil/Obinutuzumab, beides über ein Jahr gegeben, verglich (Fischer et al., Al-Sawaf et al.). Auch hier zeigte sich ein sehr ähnliches Bild, der Vorteil der zielgerichteten Therapie war bei den Hochrisikopatienten (p53-Alteration, IgHV-unmutiert) am größten, bei längerer Nachbeobachtungszeit ergab sich jedoch auch ein relevanter Vorteil für die IgHV-mutierten Patienten bei identem Gesamtüberleben. Beeindruckend war wiederum die hohe MRD-Rate von 65 % im experimentellen Arm. Venetoclax wurde folglich zusammen mit Obinutuzumab für die Erstlinienbehandlung zugelassen, überraschenderweise auch für die jungen fitten Patienten, die gar kein Teil der Stichprobe in der CLL14-Studie waren.
Zukunft
Zurzeit stehen uns in der Erstlinienbehandlung der CLL mehrere Optionen zur Verfügung (Abb. 2):
- Ibrutinib-Dauertherapie, eventuell durch Obinutuzumab ergänzt für alle Patienten
- Venetoclax/Obinutuzumab für 1 Jahr für alle Patienten
- Acalabrutinib-Dauertherapie, eventuell durch Obinutuzumab ergänzt, für alle Patienten
- Chemoimmuntherapie für Patienten ohne p53-Alteration, ohne komplexen Karyotyp und mit mutiertem IgHV-Status, wobei hier v. a. FCR für die jungen fitten Patienten relevant erscheint
Die Kombination Ibrutinib/Venetoclax wird zurzeit mit einer definiert langen Behandlungsdauer, auch MRD-getriggert in Phase-II-Studien, geprüft (Captivate, Clarity). Die deutsche CLL-Studiengruppe vergleicht zur Zeit im Rahmen des CLL17-Protokolls diese drei Strategien, nämlich Ibrutinib bis zum Progress, Venetoclax/Obinutuzumab für 1 Jahr und die Kombination Ibrutinib/Venetoclax für 12 Monate nach einer 3-monatigen Ibrutinib-Vorphase, um die Tumorlyse abzufedern. Schließlich steht die nächste Generation der Kinasehemmer kurz vor der Marktzulassung: Zanubrutinib, ein selektiverer BTK-Inhibitor, ist bereits in China und den USA verfügbar. Der nichtkovalente BTK-Inhibitor Pirtobrutinib (LOXO305) wird derzeit in Studien geprüft und ist auch bei BTK-Mutationen wie C481S wirksam. Bei Vorliegen von p53-Alterationen (Mutation oder Deletion) scheint eine dauerhafte Therapie erfolgversprechender zu sein. Moderne nebenwirkungsärmere PI3K-Inhibitoren stehen mit Duvelisib und Umbralisib in absehbarer Zeit zur Verfügung.

Resümee
Mit der Einführung der neuen zielgerichteten Substanzen, die unabhängig von ungünstigen genetischen Risikofaktoren wirksam sind, hat die Chemoimmuntherapie stark an Bedeutung verloren. Die Kombination von BTK- und BCL-2-Inhibitoren erlaubt eine definierte Therapiedauer.
Detaillierte Literatur beim Verfasser

AutorIn: OA Dr. Thomas Nösslinger
3. Medizinische Abteilung für Hämatologie und Onkologie
Hanusch-Krankenhaus, Wien

Ursprünglich erschienen:
UIM 04|2021
UIM 04|2021